

Review
doi: 10.1021/ar970004h.
De novo design of helical bundles as models for understanding protein folding and function
Affiliations
- PMID: 11087311
- PMCID: PMC3050006
- DOI: 10.1021/ar970004h
Item in Clipboard
Review
De novo design of helical bundles as models for understanding protein folding and function
Acc Chem Res.
2000 Nov.
Abstract
De novo protein design has proven to be a powerful tool for understanding protein folding, structure, and function. In this Account, we highlight aspects of our research on the design of dimeric, four-helix bundles. Dimeric, four-helix bundles are found throughout nature, and the history of their design in our laboratory illustrates our hierarchic approach to protein design. This approach has been successfully applied to create a completely native-like protein. Structural and mutational analysis allowed us to explore the determinants of native protein structure. These determinants were then applied to the design of a dinuclear metal-binding protein that can now serve as a model for this important class of proteins.
Figures

Hypothetical free energy diagram for a protein. Each line or bar represents a distinct conformational state, with the native state as the lowest energy state and the unfolded states a densely populated, nearly isoenergetic ensemble. Between these extremes are non-native folded states, oftentimes referred to as the molten globule ensemble. These non-native folded states exhibit extensive secondary structure but lack well-defined tertiary structure. The population of each state is dictated by the Boltzmann distribution. For native protein structure the free energy gap, Δ, must be large enough to significantly populate a distinct native state. For natural proteins Δ has evolved to be much larger than in de novo-designed proteins. Thus, a single mutation in designed proteins offers the advantage of being able to access the non-native states in a manner not often observed with natural proteins.

A dimeric, four-helix bundle can adopt six distinct topologies. The topology diagrams shown are for the three possible clockwise-turning helical bundles. Counterclockwise-turning helical bundles are also possible and have interfacial interactions different from their clockwise-turning correlates.

Amino acid sequences of the α2 family, reflecting the hierarchical approach to protein design. Each peptide is comprised of 35 residues with the N-terminus acetylated and the C-terminus amidated. α2B is comprised solely of leucine residues in the hydrophobic core positions. α2C is comprised of a more diverse set of nonpolar and aromatic residues in the hydrophobic core positions. α2D has three additional changes at positions 7, 26, and 30.
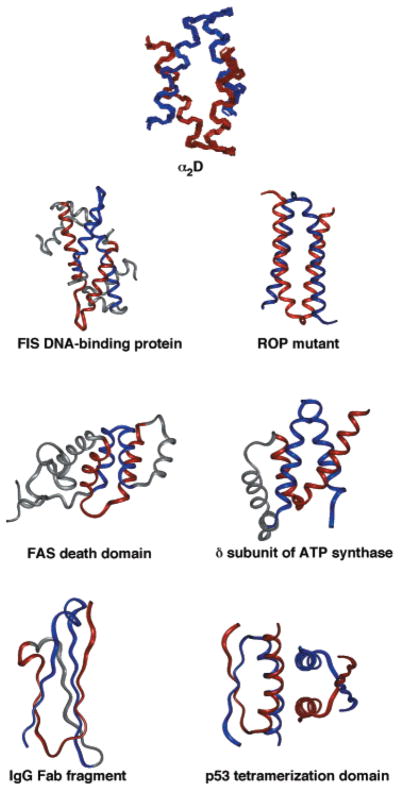
The bisecting U motif is a common structural motif. α2D was the first protein in which the bisecting U motif was recognized. (A backbone overlay of the 10 lowest energy NMR structures is shown.) Subsequently, this motif has been identified in a wide variety of proteins, including other dimeric helical bundles (FIS DNA-binding protein and Rop mutant68), intramolecularly folded helical bundles (FAS death domain and N-terminal domain of the δ subunit of the ATPsynthase118), and β-sheet proteins (the IgG fold119), and multimeric proteins with both α/β structures (p53 tetramerization domain120).

α2D has three nonequivalent interfaces: (a) Interdigitated Leu residues stabilize the interface between helices 1 and 1′. (b) A diverse collection of aromatic and hydrophobic residues stabilize the interface between helices 1 and 2′. (c) Hydrogen-bonded clusters of His residues stabilize the interface between helices 2 and 2′.

Glu7 is not involved in stabilizing the native state of α2D. Superposition of the 10 lowest energy NMR-derived structures (left panel) shows that Glu7 adopts multiple conformations typical of solvent-exposed residues (hydrophobic core residues in gray). Replacing Glu7 with other polar or nonpolar residues has little effect on the overall stability (ΔG) of the protein but has a large effect on the conformational specificity (Δ) of the protein (right panel). For example, replacing Glu7 with a polar residue (as in E7H) results in a protein with a Δ large enough to adopt a native structure. However, replacing Glu7 with a hydrophobic residue (as in E7V) results in a protein that has lost its ability to adopt a native structure. Note that the number of conformations that E7V adopts cannot be too large because this would result in a large entropic stabilization of this ensemble, which is not observed. In conclusion, Glu7 is involved in “negative design” by increasing Δ through the destabilization of the non-native folded states relative to the native state. We assume that the energy level of the unfolded ensemble is not significantly affected by these substitutions.

Hydrogen-bonding within a His cluster is essential for the native state of α2D. An interior His (H26) forms a hydrogen bond across the interface with an exterior His (H30′) on the other monomer (left panel). A symmetrical interaction also occurs between H26′ and H30. Substitution of residues that are unable to form hydrogen bonds (as in H30K) results in a protein that has lost all ability to adopt the native structure. Substitution of residues that are capable of forming hydrogen bonds (as in H30D) results in a protein that maintains the native structure (right panel). Note that the non-native states of H30K are depicted slightly lower in energy than those of α2D because Lys is known have a higher helical propensity than His. In conclusion, the His cluster is involved in “positive design” by increasing Δ through the direct stabilization of the native state relative to the non-native folded states.
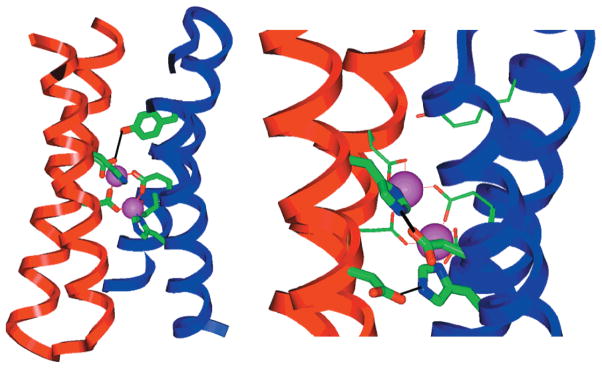
X-ray structure of the di-Zn(II) form of DF1 (2.5 Å resolution), which is nearly identical to the intended design. The backbone of the structure plus the ligands are shown in the two views. At left, a Tyr phenolic group hydrogen bonds to a Glu carboxylate (a second, symmetry-related Tyr-Glu interaction is not shown for clarity). At right, hydrogen bonds between Asp carboxylates and His side chains are shown.
References
-
- Cordes MHJ, Davidson AR, Sauer RT. Sequence space, folding and protein design. Curr Opin Struct Biol. 1996;6:3–10. - PubMed
-
- Dill KA. Dominant forces in protein folding. Biochemistry. 1990;31:7133–7155. - PubMed
-
- Dobson CM. Characterization of protein folding intermediates. Curr Opin Struct Biol. 1991;1:22–27.
-
- Eaton WA, Thompson PA, Chan CK, Hagen SJ, Hofrichter J. Fast events in protein folding. Structure. 1996;4:1133–1139. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
