

3' deletions cause aniridia by preventing PAX6 gene expression
- PMID: 11087823
- PMCID: PMC17648
- DOI: 10.1073/pnas.240398797
3' deletions cause aniridia by preventing PAX6 gene expression
Abstract
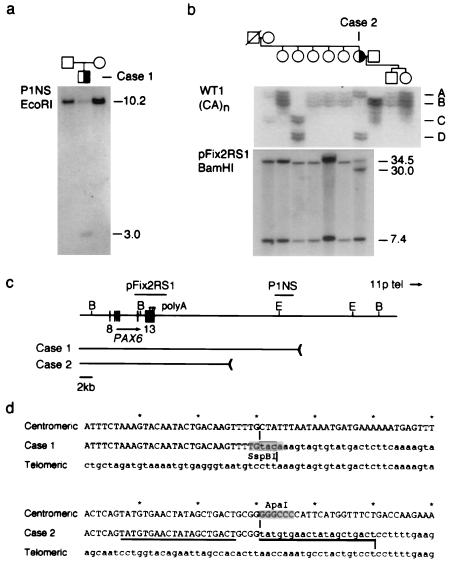
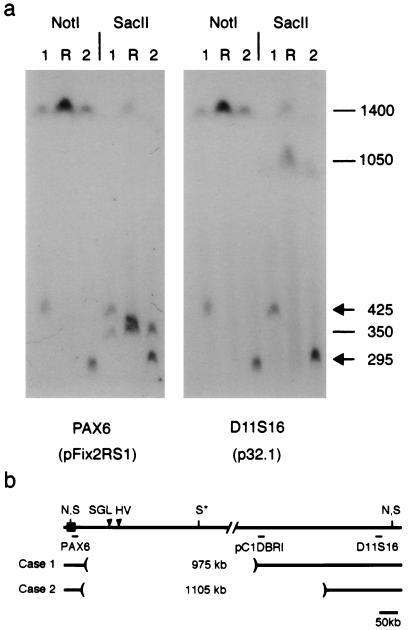
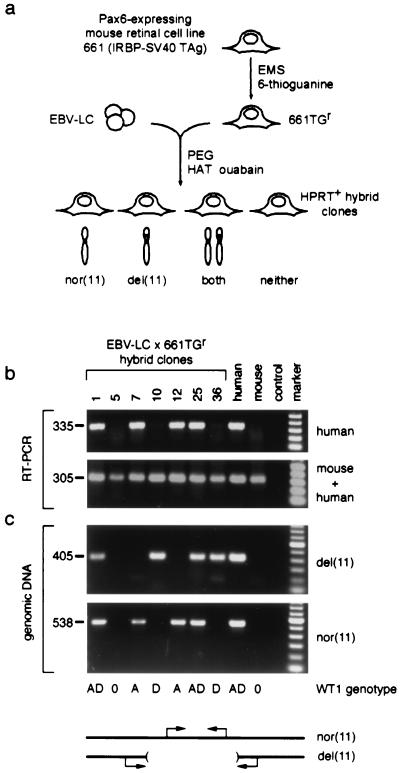
Aniridia is a panocular human eye malformation caused by heterozygous null mutations within PAX6, a paired-box transcription factor, or cytogenetic deletions of chromosome 11p13 that encompass PAX6. Chromosomal rearrangements also have been described that disrupt 11p13 but spare the PAX6 transcription unit in two families with aniridia. These presumably cause a loss of gene expression, by removing positive cis regulatory elements or juxtaposing negative DNA sequences. We report two submicroscopic de novo deletions of 11p13 that cause aniridia but are located >11 kb from the 3' end of PAX6. The clinical manifestations are indistinguishable from cases with chain-terminating mutations in the coding region. Using human x mouse retinoblastoma somatic cell hybrids, we show that PAX6 is transcribed only from the normal allele but not from the deleted chromosome 11 homolog. Our findings suggest that remote 3' regulatory elements are required for initiation of PAX6 expression.
Figures
References
-
- Gehring W J, Ikeo K. Trends Genet. 1999;15:371–377. - PubMed
-
- Ton C C, Hirvonen H, Miwa H, Weil M M, Monaghan P, Jordan T, van Heyningen V, Hastie N D, Meijers-Heijboer H, Drechsler M, et al. Cell. 1991;67:1059–1074. - PubMed
-
- Hanson I, Jordan T, Van Heyningen V. In: Molecular Genetics of Inherited Eye Disorders. Wright A F, Jay B, editors. Switzerland: Harwood Academic; 1994. pp. 445–467.
-
- Prosser J, van Heyningen V. Hum Mutat. 1998;11:93–108. - PubMed
-
- Glaser T, Walton D S, Maas R L. Nat Genet. 1992;2:232–239. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
