

Review
doi: 10.1186/ar73.
Epub 2000 Feb 24.
A revival of the B cell paradigm for rheumatoid arthritis pathogenesis?
Affiliations
- PMID: 11094418
- PMCID: PMC129991
- DOI: 10.1186/ar73
Item in Clipboard
Review
A revival of the B cell paradigm for rheumatoid arthritis pathogenesis?
Arthritis Res.
2000.
Abstract
Dominant paradigms for the understanding of rheumatoid arthritis (RA) pathogenesis have changed over the years. A predominant role of B lymphocytes, and perhaps of the rheumatoid factor they produced, was initially invoked. In more recent years, recognition of antigens in the joint by T cells sparking an inflammatory cascade has been a more favored interpretation. Here, we re-examine some of the arguments that underpin this proposed role of joint T cells, in light of recent results from transgenic mice in which a self-reactive T-cell receptor provokes disease, but from outside the joint and indirectly via B lymphocytes and immunoglobulins.
Figures
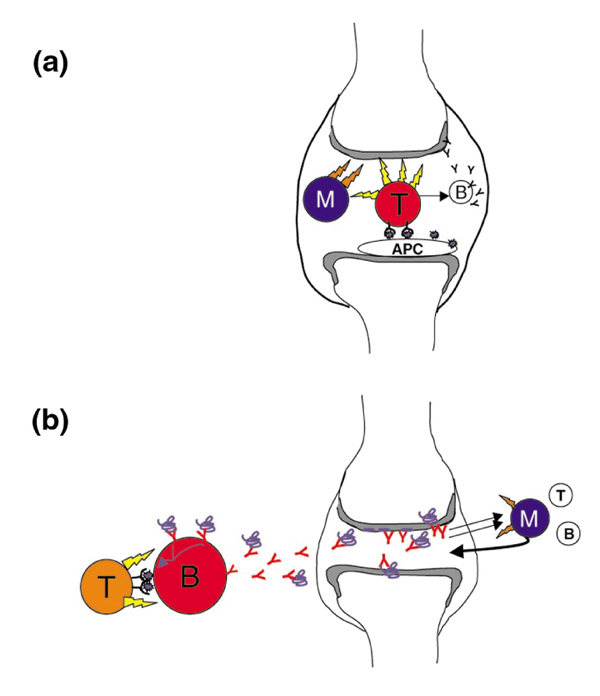
Models of T cell invovement in rheumatoid arthritis (RA). (a) The 'classical' model. T cells within the joint recognize fragments of autoantigens presented by local dendritic cells (DCs). As a consequence they produce inflammatory cytokines that directly affect chondrocytes, but mainly prime monocytes and synoviocytes (either fibroblastic or macrophage-like) to produce more substantial amounts of monokines (eg tumor necrosis factor), proteases or glycosidases to effect cartilage destruction and bone remodeling. In this view, there may be local activation of B cells to produce immunoglobulins directed against joint-specific structures, but this is an ancillary pathway. (b) In this model, T-cell activation occurs outside the joint, and is not necessarily caused by joint-specific peptides. The relevant mode of antigen presentation is by B cells, which have picked up rare antigen molecules via their surface immunoglobulin, and are thus preferentially helped. The arthritogenic immunoglobulin they produce (in isolation or in complexed form) then diffuses to the joint. In this locale, immunoglobulin binding or activation by immune complexes provokes the release of chemokines, attracting and priming monocytes to elicit cartilage and joint destruction (and secondary release of B and T cells).
Comment in
-
Rheumatoid arthritis viewed using a headache paradigm.Arthritis Res. 2000;2(3):169-71. doi: 10.1186/ar84. Epub 2000 Apr 6. Arthritis Res. 2000. PMID: 11094426 Free PMC article. Review.
References
-
- Zvaifler NJ. The immunopathology of joint inflammation in rheumatoid arthritis. Adv Immunol. 1973;265:265–336. - PubMed
-
- Ohno O, Cooke D. Electron microscopic morphology of immunoglobulin aggregates and their interactions in rheumatoid articular collagenous tissues. Arthritis Rheum. 1978;21:516–527. - PubMed
-
- Vaughan JH. Pathogenic concepts and origins of rheumatoid factor in rheumatoid arthritis. Arthritis Rheum. 1993;36:1–6. - PubMed
-
- Smolen JS, Steiner G. Are autoantibodies active players or epiphenomena? Curr Opin Rheumatol. 1998;10:201–206. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
