

Presenilin-1 P264L knock-in mutation: differential effects on abeta production, amyloid deposition, and neuronal vulnerability
- PMID: 11102478
- PMCID: PMC6773081
- DOI: 10.1523/JNEUROSCI.20-23-08717.2000
Presenilin-1 P264L knock-in mutation: differential effects on abeta production, amyloid deposition, and neuronal vulnerability
Abstract
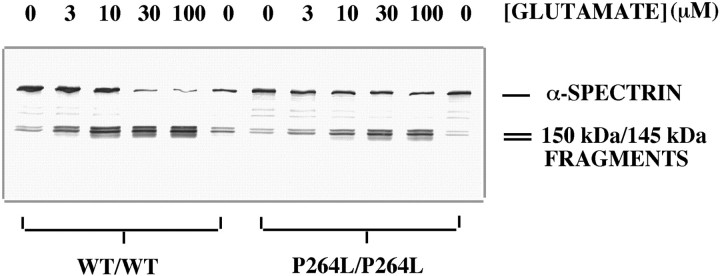
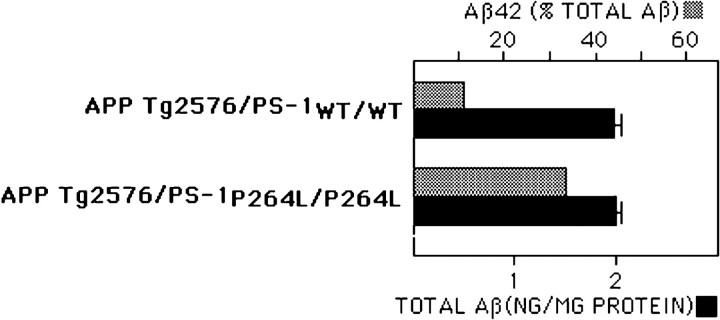
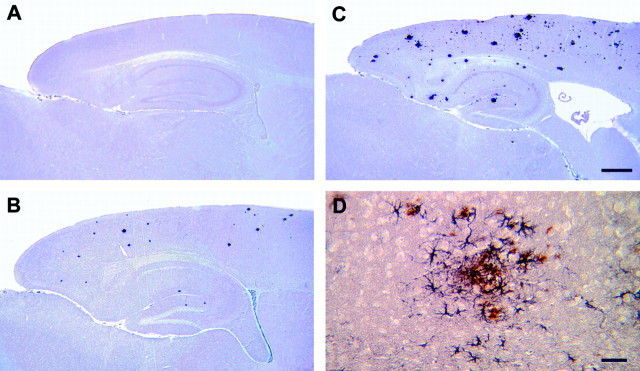
The pathogenic mechanism linking presenilin-1 (PS-1) gene mutations to familial Alzheimer's disease (FAD) is uncertain, but has been proposed to include increased neuronal sensitivity to degeneration and enhanced amyloidogenic processing of the beta-amyloid precursor protein (APP). We investigated this issue by using gene targeting with the Cre-lox system to introduce an FAD-linked P264L mutation into the endogenous mouse PS-1 gene, an approach that maintains normal regulatory controls over expression. Primary cortical neurons derived from PS-1 homozygous mutant knock-in mice exhibit basal neurodegeneration similar to their PS-1 wild-type counterparts. Staurosporine and Abeta1-42 induce apoptosis, and neither the dose dependence nor maximal extent of cell death is altered by the PS-1 knock-in mutation. Similarly, glutamate-induced neuronal necrosis is unaffected by the PS-1P264L mutation. The lack of effect of the PS-1P264L mutation is confirmed by measures of basal- and toxin-induced caspase and calpain activation, biochemical indices of apoptotic and necrotic signaling, respectively. To analyze the influence of the PS-1P264L knock-in mutation on APP processing and the development of AD-type neuropathology, we created mouse lines carrying mutations in both PS-1 and APP. In contrast to the lack of effect on neuronal vulnerability, cortical neurons cultured from PS-1P264L homozygous mutant mice secrete Abeta42 at an increased rate, whereas secretion of Abeta40 is reduced. Moreover, the PS-1 knock-in mutation selectively increases Abeta42 levels in the mouse brain and accelerates the onset of amyloid deposition and its attendant reactive gliosis, even as a single mutant allele. We conclude that expression of an FAD-linked mutant PS-1 at normal levels does not generally increase cortical neuronal sensitivity to degeneration. Instead, enhanced amyloidogenic processing of APP likely is critical to the pathogenesis of PS-1-linked FAD.
Figures
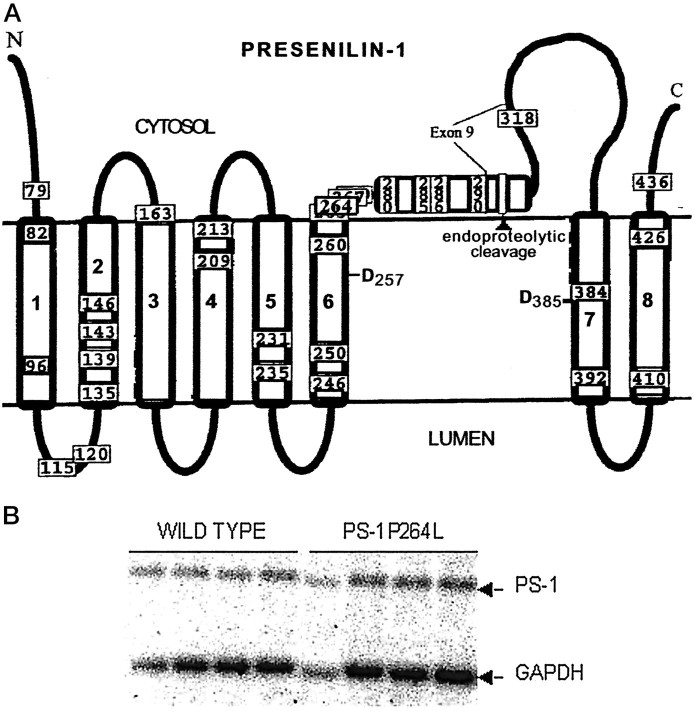
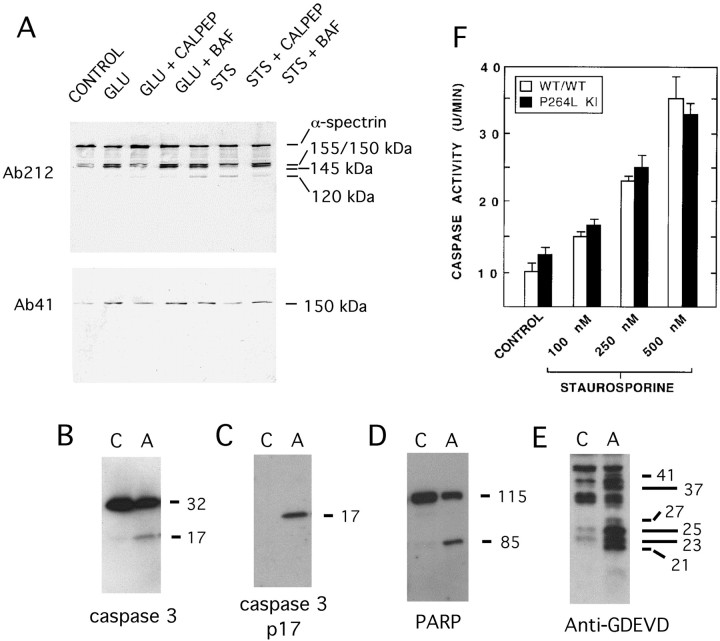
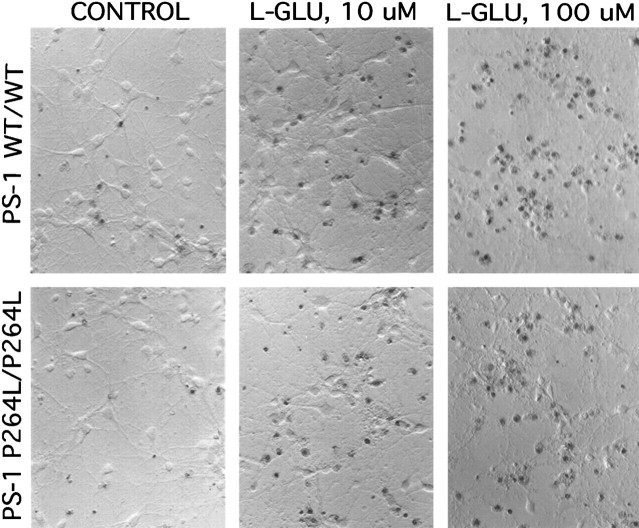
References
-
- Banker G, Goslin K. Culturing nerve cells. MIT; Cambridge, MA: 1998.
-
- Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitski T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer's disease-linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. - PubMed
-
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. - PubMed
-
- Cai XD, Golde TE, Younkin SG. Release of excess amyloid β protein from a mutant amyloid β-protein precursor. Science. 1993;259:514–516. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases