

An abnormal Ca(2+) response in mutant sarcomere protein-mediated familial hypertrophic cardiomyopathy
- PMID: 11104788
- PMCID: PMC381468
- DOI: 10.1172/JCI11093
An abnormal Ca(2+) response in mutant sarcomere protein-mediated familial hypertrophic cardiomyopathy
Abstract
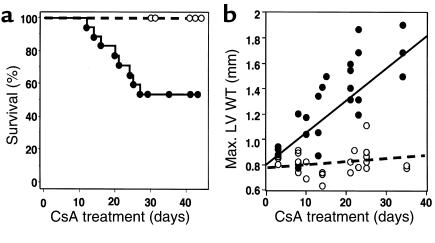
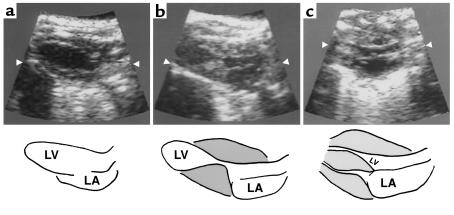
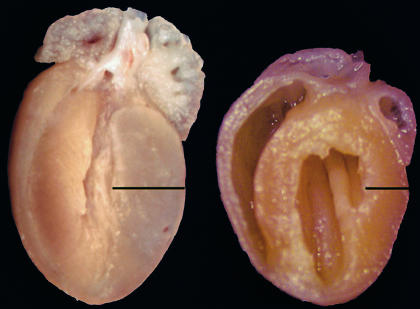
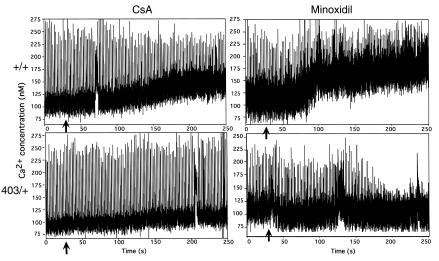
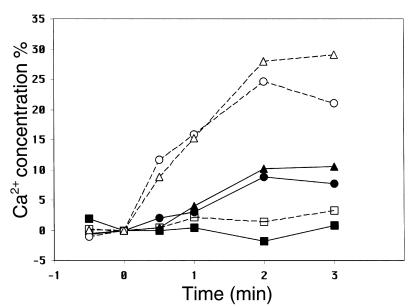
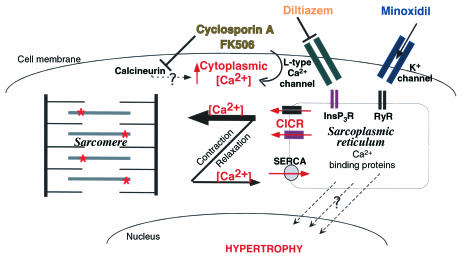
Dominant-negative sarcomere protein gene mutations cause familial hypertrophic cardiomyopathy (FHC), a disease characterized by left-ventricular hypertrophy, angina, and dyspnea that can result in sudden death. We report here that a murine model of FHC bearing a cardiac myosin heavy-chain gene missense mutation (alphaMHC(403/+)), when treated with calcineurin inhibitors or a K(+)-channel agonist, developed accentuated hypertrophy, worsened histopathology, and was at risk for early death. Despite distinct pharmacologic targets, each agent augmented diastolic Ca(2+) concentrations in wild-type cardiac myocytes; alphaMHC(403/+) myocytes failed to respond. Pretreatment with a Ca(2+)-channel antagonist abrogated diastolic Ca(2+) changes in wild-type myocytes and prevented the exaggerated hypertrophic response of treated alphaMHC(403/+) mice. We conclude that FHC-causing sarcomere protein gene mutations cause abnormal Ca(2+) responses that initiate a hypertrophic response. These data define an important Ca(2+)-dependent step in the pathway by which mutant sarcomere proteins trigger myocyte growth and remodel the heart, provide definitive evidence that environment influences progression of FHC, and suggest a rational therapeutic approach to this prevalent human disease.
Figures

Comment in
-
Making matters worse for a broken heart.J Clin Invest. 2000 Dec;106(12):1437-9. doi: 10.1172/JCI11733. J Clin Invest. 2000. PMID: 11120749 Free PMC article. No abstract available.
References
-
- Maron BJ, et al. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92:785–789. - PubMed
-
- Colluci, W.S., and Braunwald, E.B. 1997. Pathophysiology of heart failure. In Heart disease. E. Braunwald, editor. W.B. Saunders Co. Philadelphia, Pennsylvania, USA. 394–420.
-
- Benjamin EJ, Levy D. Why is left ventricular hypertrophy so predictive of morbidity and mortality? Am J Med Sci. 1999;317:168–175. - PubMed
-
- Fatkin, D., Seidman, J.G., and Seidman, C.E. 2000. Hypertrophic cardiomyopathy. In Cardiovascular medicine. J.T. Willerson and J.N. Cohn, editors. W.B. Saunders Co. Philadelphia, Pennsylvania, USA. 1055–1075.
-
- Hunter, J.J., Grace, A., and Chien, K.R. 1999. Molecular and cellular biology of cardiac hypertrophy and failure. In Molecular basis of cardiovascular disease. K.R. Chien, editor. W.B. Saunders Co. Philadelphia, Pennsylvania, USA. 211–250.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
