

Biochemical and biological functions of the N-terminal, noncatalytic domain of extracellular signal-regulated kinase 2
- PMID: 11113199
- PMCID: PMC88798
- DOI: 10.1128/MCB.21.1.249-259.2001
Biochemical and biological functions of the N-terminal, noncatalytic domain of extracellular signal-regulated kinase 2
Abstract
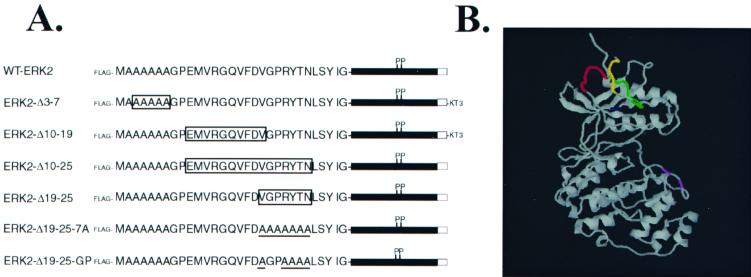
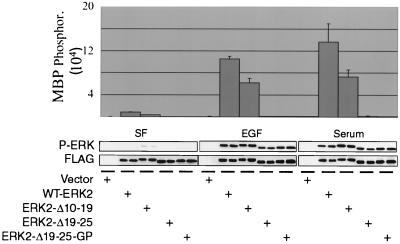
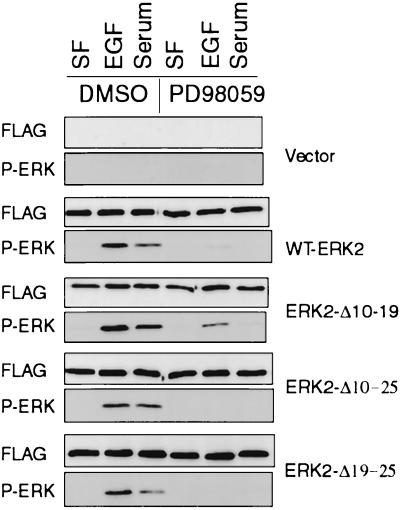
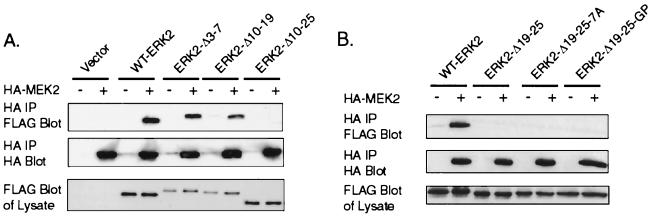
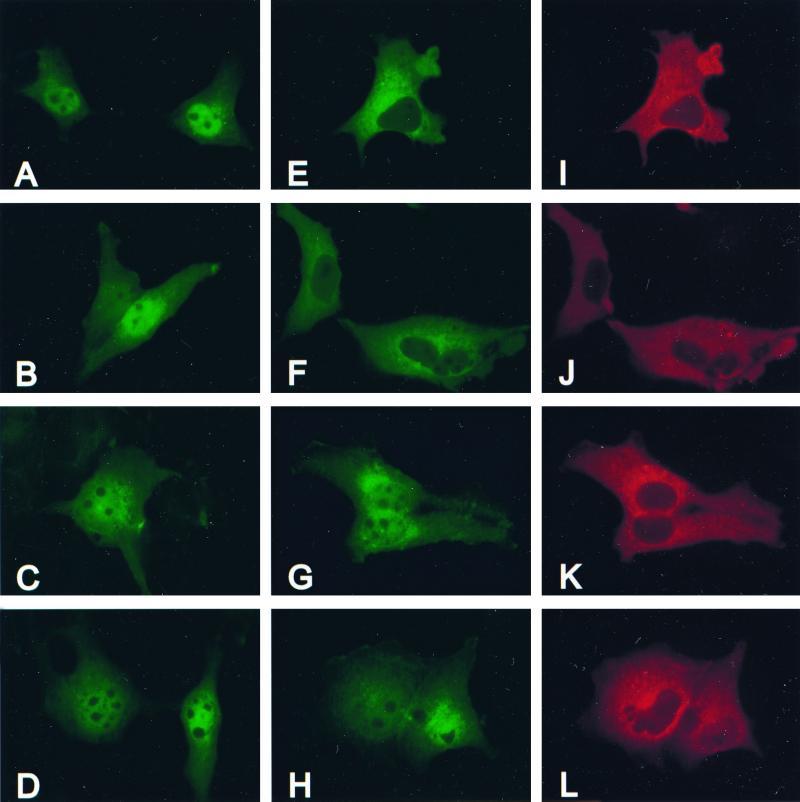
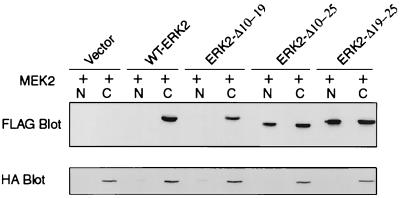
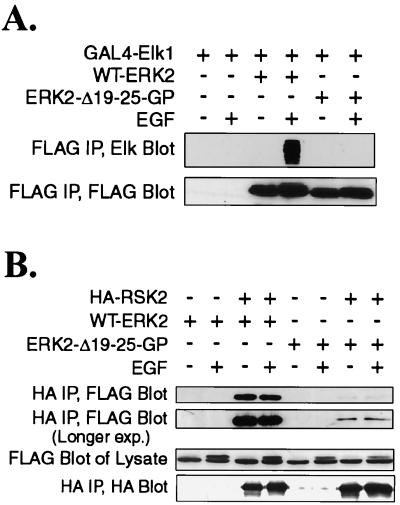
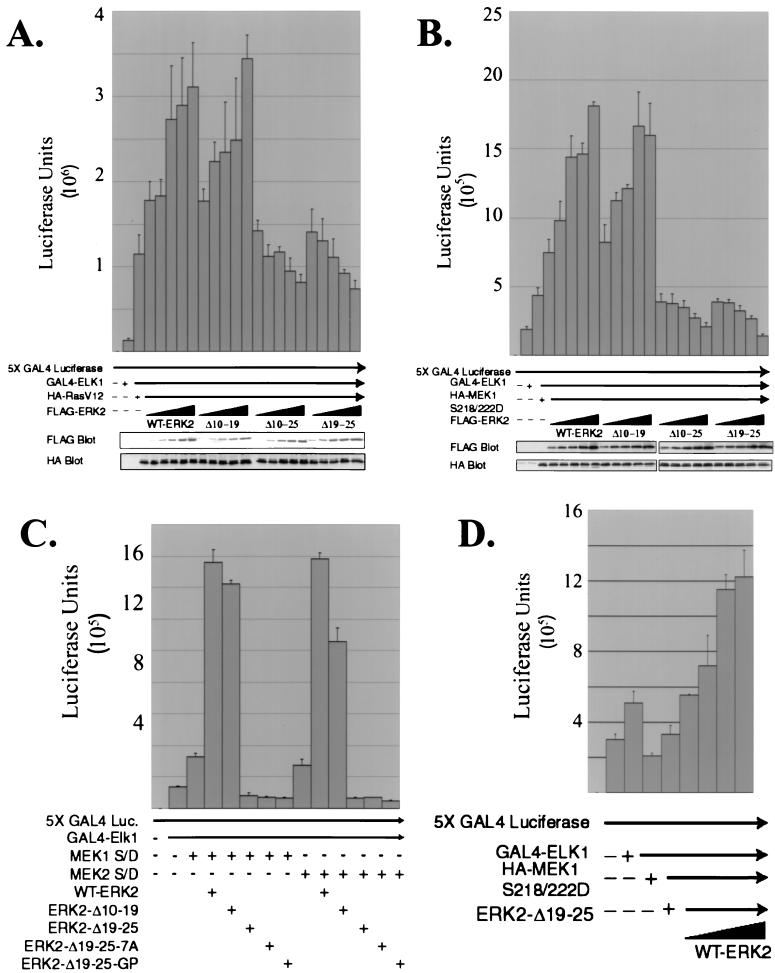
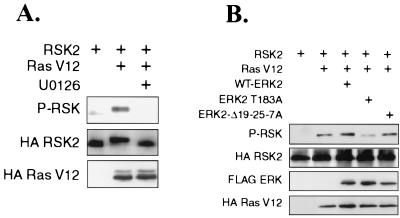
Extracellular signal-regulated kinase 1 (ERK1) and ERK2 are important components in signal transduction pathways involved in many cellular processes, including cell differentiation and proliferation. These proteins consist of a central kinase domain flanked by short N- and C-terminal noncatalytic domains. While the regulation of ERK2 by sequences within the kinase domain has been extensively studied, little is known about the small regions outside of the kinase domain. We performed mutational analysis on the N-terminal, noncatalytic domain of ERK2 in an attempt to determine its role in ERK2 function and regulation. Deleting or mutating amino acids 19 to 25 (ERK2-Delta19-25) created an ERK2 molecule that could be phosphorylated in response to growth factor and serum stimulation in a MEK (mitogen-activated protein kinase kinase or ERK kinase)-dependent manner but had little kinase activity and was unable to bind to MEK in vivo. Since MEK acts as a cytoplasmic anchor for the ERKs, the lack of a MEK interaction resulted in the aberrant nuclear localization of ERK2-Delta19-25 mutants in serum-starved cells. Assaying these mutants for their ability to affect ERK signaling revealed that ERK2-Delta19-25 mutants acted in a dominant-negative manner to inhibit transcriptional signaling through endogenous ERKs to an Elk1-responsive promoter in transfected COS-1 cells. However, ERK2-Delta19-25 had no effect on the phosphorylation of RSK2, an ERK2 cytoplasmic substrate, whereas a nonactivatable ERK (T183A) that retained these sequences could inhibit RSK2 phosphorylation. These results suggest that the N-terminal domain of ERK2 profoundly affects ERK2 localization, MEK binding, kinase activity, and signaling and identify a novel dominant-negative mutant of ERK2 that can dissociate at least some transcriptional responses from cytoplasmic responses.
Figures
Similar articles
-
Rac-PAK signaling stimulates extracellular signal-regulated kinase (ERK) activation by regulating formation of MEK1-ERK complexes.Mol Cell Biol. 2002 Sep;22(17):6023-33. doi: 10.1128/MCB.22.17.6023-6033.2002. Mol Cell Biol. 2002. PMID: 12167697 Free PMC article.
-
Docking sites on mitogen-activated protein kinase (MAPK) kinases, MAPK phosphatases and the Elk-1 transcription factor compete for MAPK binding and are crucial for enzymic activity.Biochem J. 2003 Mar 15;370(Pt 3):1077-85. doi: 10.1042/BJ20021806. Biochem J. 2003. PMID: 12529172 Free PMC article.
-
Identifying specific kinase substrates through engineered kinases and ATP analogs.Methods. 2004 Apr;32(4):389-97. doi: 10.1016/j.ymeth.2003.10.002. Methods. 2004. PMID: 15003601
-
Dimerization in MAP-kinase signaling.Trends Biochem Sci. 2000 Jan;25(1):7-9. doi: 10.1016/s0968-0004(99)01508-x. Trends Biochem Sci. 2000. PMID: 10637602 Review.
-
A kinetic approach towards understanding substrate interactions and the catalytic mechanism of the serine/threonine protein kinase ERK2: identifying a potential regulatory role for divalent magnesium.Biochim Biophys Acta. 2004 Mar 11;1697(1-2):81-7. doi: 10.1016/j.bbapap.2003.11.015. Biochim Biophys Acta. 2004. PMID: 15023352 Review.
Cited by
-
Monomeric and dimeric models of ERK2 in conjunction with studies on cellular localization, nuclear translocation, and in vitro analysis.Mol Cells. 2012 Apr;33(4):325-34. doi: 10.1007/s10059-012-0023-4. Epub 2012 Mar 23. Mol Cells. 2012. PMID: 22450690 Free PMC article. Review.
-
Signalling specificity of Ser/Thr protein kinases through docking-site-mediated interactions.Biochem J. 2003 May 15;372(Pt 1):1-13. doi: 10.1042/BJ20021641. Biochem J. 2003. PMID: 12600273 Free PMC article. Review.
-
EDD enhances cell survival and cisplatin resistance and is a therapeutic target for epithelial ovarian cancer.Carcinogenesis. 2014 May;35(5):1100-9. doi: 10.1093/carcin/bgt489. Epub 2013 Dec 30. Carcinogenesis. 2014. PMID: 24379240 Free PMC article.
-
Alteration of Akt activity increases chemotherapeutic drug and hormonal resistance in breast cancer yet confers an achilles heel by sensitization to targeted therapy.Adv Enzyme Regul. 2008;48:113-35. doi: 10.1016/j.advenzreg.2008.02.006. Epub 2008 Feb 21. Adv Enzyme Regul. 2008. PMID: 18423407 Free PMC article.
-
ERK2-topoisomerase II regulatory axis is important for gene activation in immediate early genes.Nat Commun. 2023 Dec 14;14(1):8341. doi: 10.1038/s41467-023-44089-y. Nat Commun. 2023. PMID: 38097570 Free PMC article.
References
-
- Alessi D R, Cuenda A, Cohen P, Dudley D T, Saltiel A R. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. - PubMed
-
- Brunet A, Pouyssegur J. Identification of MAP kinase domains by redirecting stress signals into growth factor responses. Science. 1996;272:1652–1655. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous