

BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension
- PMID: 11115378
- PMCID: PMC1234937
- DOI: 10.1086/316947
BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension
Abstract
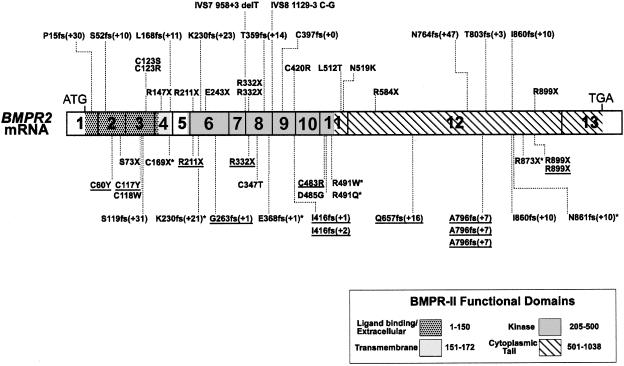
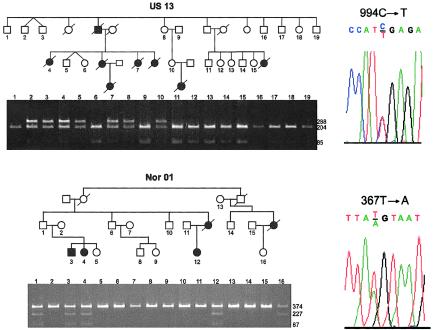
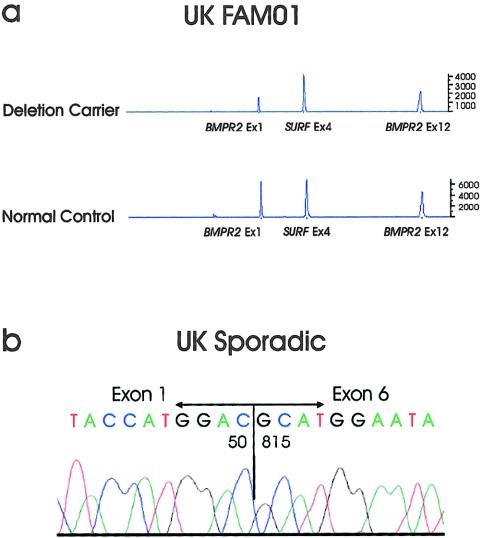
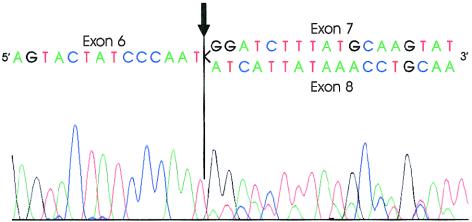
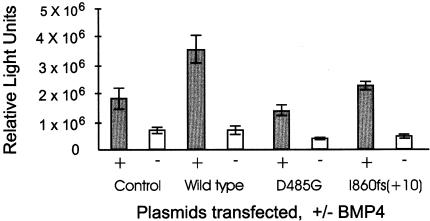
Primary pulmonary hypertension (PPH) is a potentially lethal disorder, because the elevation of the pulmonary arterial pressure may result in right-heart failure. Histologically, the disorder is characterized by proliferation of pulmonary-artery smooth muscle and endothelial cells, by intimal hyperplasia, and by in situ thrombus formation. Heterozygous mutations within the bone morphogenetic protein type II receptor (BMPR-II) gene (BMPR2), of the transforming growth factor beta (TGF-beta) cell-signaling superfamily, have been identified in familial and sporadic cases of PPH. We report the molecular spectrum of BMPR2 mutations in 47 additional families with PPH and in three patients with sporadic PPH. Among the cohort of patients, we have identified 22 novel mutations, including 4 partial deletions, distributed throughout the BMPR2 gene. The majority (58%) of mutations are predicted to lead to a premature termination codon. We have also investigated the functional impact and genotype-phenotype relationships, to elucidate the mechanisms contributing to pathogenesis of this important vascular disease. In vitro expression analysis demonstrated loss of BMPR-II function for a number of the identified mutations. These data support the suggestion that haploinsufficiency represents the common molecular mechanism in PPH. Marked variability of the age at onset of disease was observed both within and between families. Taken together, these studies illustrate the considerable heterogeneity of BMPR2 mutations that cause PPH, and they strongly suggest that additional factors, genetic and/or environmental, may be required for the development of the clinical phenotype.
Figures
References
Electronic-Database Information
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for familial PPH [MIM 178600] and hereditary hemorrhagic telangiectasia [MIM 187300 and 600376])
References
-
- Abenhaim L, Moride Y, Brenot F, Rich S, Benichou J, Kurz X, Higenbottam T, Oakley C, Wouters E, Aubier M, Simonneau G, Begaud B (1996) Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N Engl J Med 335:609–616 - PubMed
-
- Deng Z, Haghighi F, Helleby L, Vanterpool K, Horn EM, Barst RJ, Hodge SE, Morse JH, Knowles JA (2000a) Fine mapping of PPH1, a gene for familial primary pulmonary hypertension, to a 3-cM region on chromosome 2q33. Am J Respir Crit Care Med 161:1055–1059 - PubMed
-
- Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA (2000b) Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 67:737–744 - PMC - PubMed
-
- Gaine SP, Rubin LJ (1998) Primary pulmonary hypertension. Lancet 352:719–725 - PubMed
-
- Gomez-Sanchez MA, de la Saenz C, Gomez-Pajuelo C, Martinez-Tello FJ, Mestre de Juan MJ, James TN (1991) Clinical and pathologic manifestations of pulmonary vascular disease in the toxic oil syndrome. J Am Coll Cardiol 18:1539–1545 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
