

Multifaceted regulation of cell cycle progression by estrogen: regulation of Cdk inhibitors and Cdc25A independent of cyclin D1-Cdk4 function
- PMID: 11154267
- PMCID: PMC86671
- DOI: 10.1128/MCB.21.3.794-810.2001
Multifaceted regulation of cell cycle progression by estrogen: regulation of Cdk inhibitors and Cdc25A independent of cyclin D1-Cdk4 function
Abstract
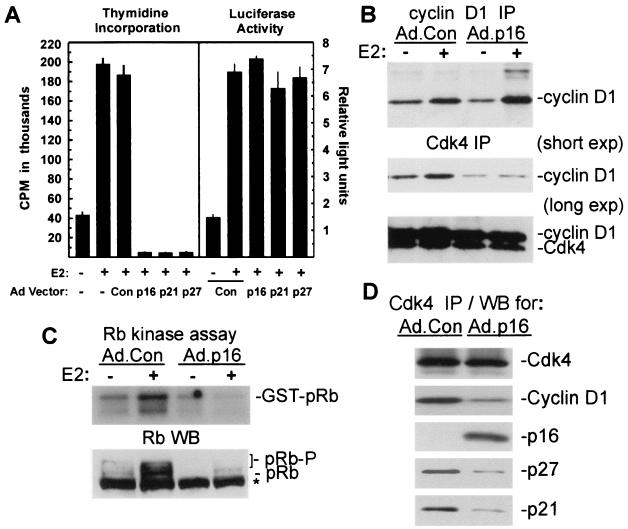
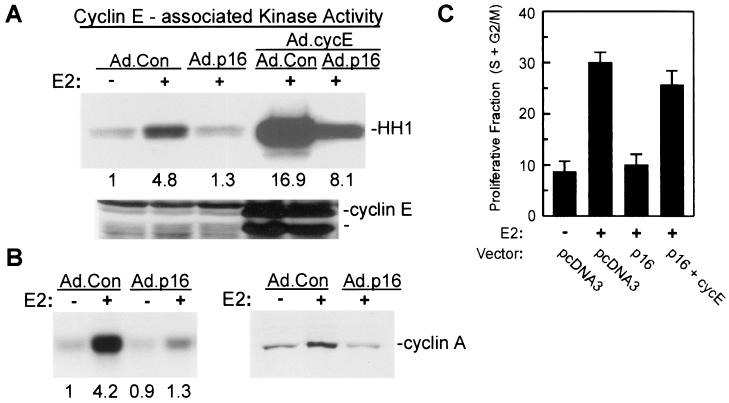
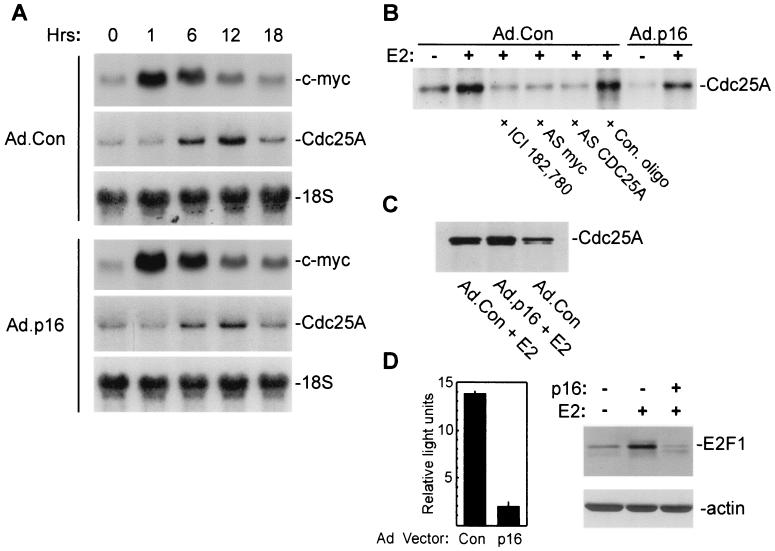
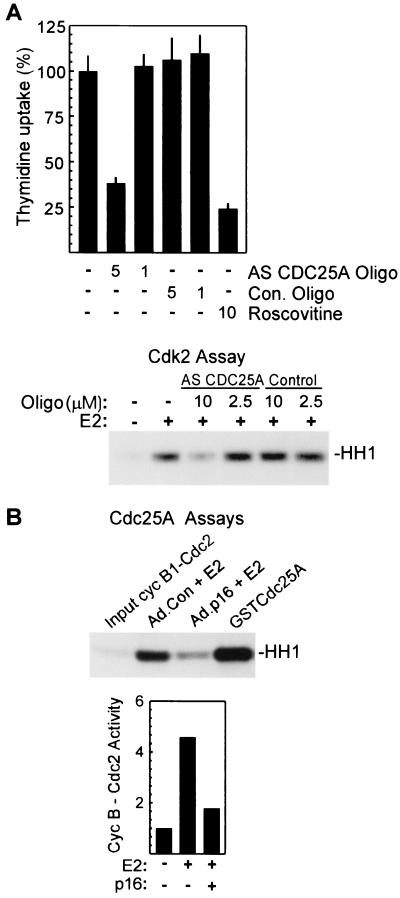
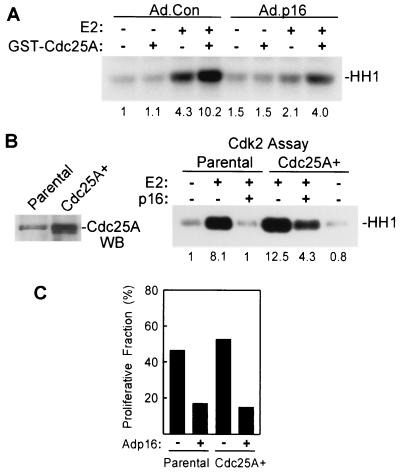
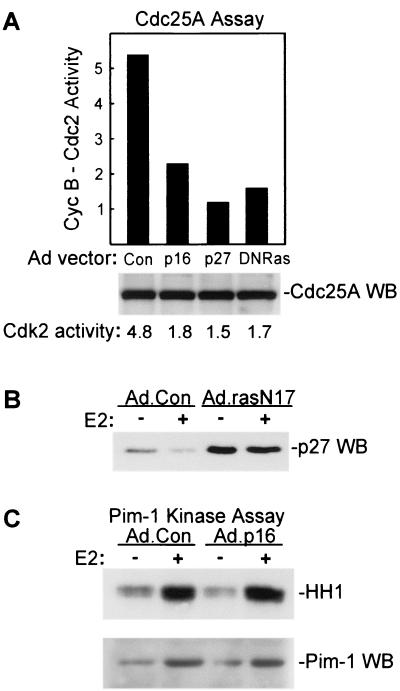
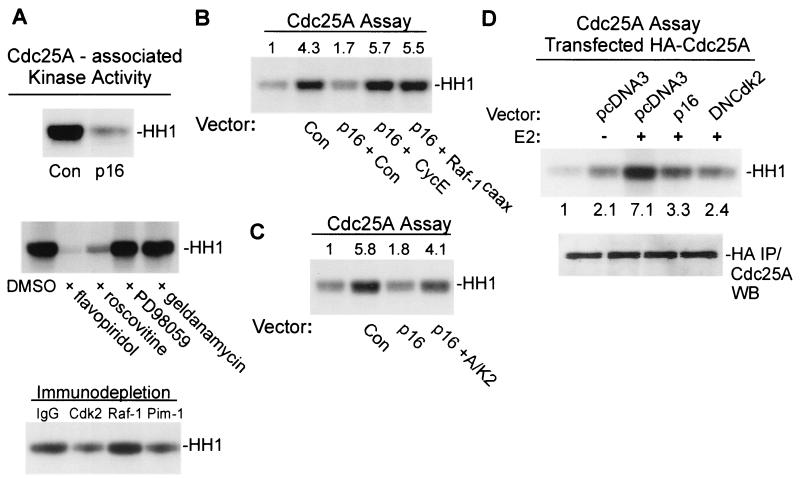
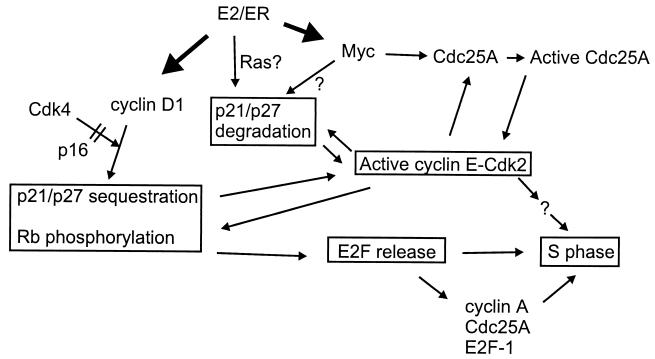
Estrogens induce proliferation of estrogen receptor (ER)-positive MCF-7 breast cancer cells by stimulating G(1)/S transition associated with increased cyclin D1 expression, activation of cyclin-dependent kinases (Cdks), and phosphorylation of the retinoblastoma protein (pRb). We have utilized blockade of cyclin D1-Cdk4 complex formation through adenovirus-mediated expression of p16(INK4a) to demonstrate that estrogen regulates Cdk inhibitor expression and expression of the Cdk-activating phosphatase Cdc25A independent of cyclin D1-Cdk4 function and cell cycle progression. Expression of p16(INK4a) inhibited G(1)/S transition induced in MCF-7 cells by 17-beta-estradiol (E(2)) with associated inhibition of both Cdk4- and Cdk2-associated kinase activities. Inhibition of Cdk2 activity was associated with delayed removal of Cdk-inhibitory activity in early G(1) and decreased cyclin A expression. Cdk-inhibitory activity and expression of both p21(Cip1) and p27(Kip1) was decreased, however, in both control and p16(INK4a)-expressing cells 20 h after estrogen treatment. Expression of Cdc25A mRNA and protein was induced by E(2) in control and p16(INK4a)-expressing MCF-7 cells; however, functional activity of Cdc25A was inhibited in cells expressing p16(INK4a). Inhibition of Cdc25A activity in p16(INK4a)-expressing cells was associated with depressed Cdk2 activity and was reversed in vivo and in vitro by active Cdk2. Transfection of MCF-7 cells with a dominant-negative Cdk2 construct inhibited the E(2)-dependent activation of ectopic Cdc25A. Supporting a role for Cdc25A in estrogen action, antisense CDC25A oligonucleotides inhibited estrogen-induced Cdk2 activation and DNA synthesis. In addition, inactive cyclin E-Cdk2 complexes from p16(INK4a)-expressing, estrogen-treated cells were activated in vitro by treatment with recombinant Cdc25A and in vivo in cells overexpressing Cdc25A. The results demonstrate that functional association of cyclin D1-Cdk4 complexes is required for Cdk2 activation in MCF-7 cells and that Cdk2 activity is, in turn, required for the in vivo activation of Cdc25A. These studies establish Cdc25A as a growth-promoting target of estrogen action and further indicate that estrogens independently regulate multiple components of the cell cycle machinery, including expression of p21(Cip1) and p27(Kip1).
Figures

References
-
- Altucci L, Addeo R, Cicatiello L, Dauvois S, Parker M G, Truss M, Beato M, Sica V, Bresciani F, Weisz A. 17beta-Estradiol induces cyclin D1 gene transcription, p36D1–p34cdk4 complex activation and p105Rb phosphorylation during mitogenic stimulation of G(1)-arrested human breast cancer cells. Oncogene. 1996;12:2315–2324. - PubMed
-
- Altucci L, Addeo R, Cicatiello L, Germano D, Pacilio C, Battista T, Cancemi M, Petrizzi V B, Bresciani F, Weisz A. Estrogen induces early and timed activation of cyclin-dependent kinases 4, 5, and 6 and increases cyclin messenger ribonucleic acid expression in rat uterus. Endocrinology. 1997;138:978–984. - PubMed
-
- Brenner A J, Stampfer M R, Aldaz C M. Increased p16 expression with first senescence arrest in human mammary epithelial cells and extended growth capacity with p16 inactivation. Oncogene. 1998;17:199–205. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous