

Efficiency of database search for identification of mutated and modified proteins via mass spectrometry
- PMID: 11157792
- PMCID: PMC544186
- DOI: 10.1101/gr.154101
Efficiency of database search for identification of mutated and modified proteins via mass spectrometry
Abstract
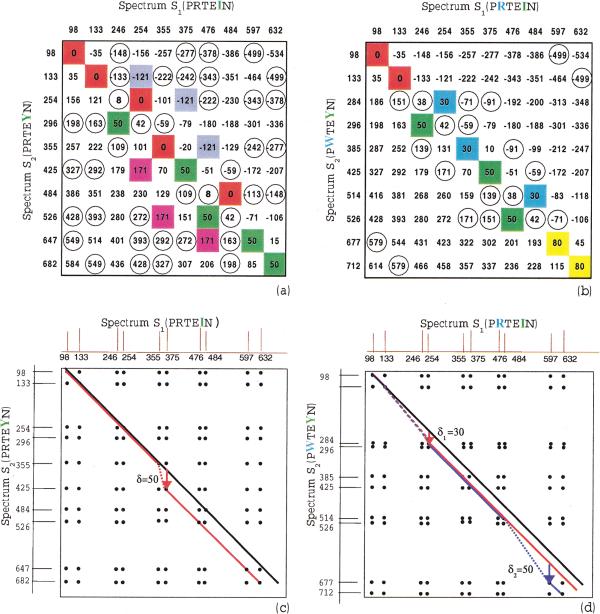
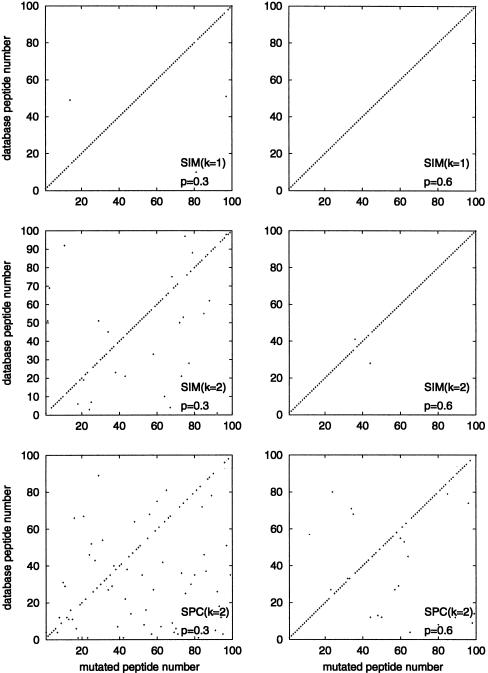
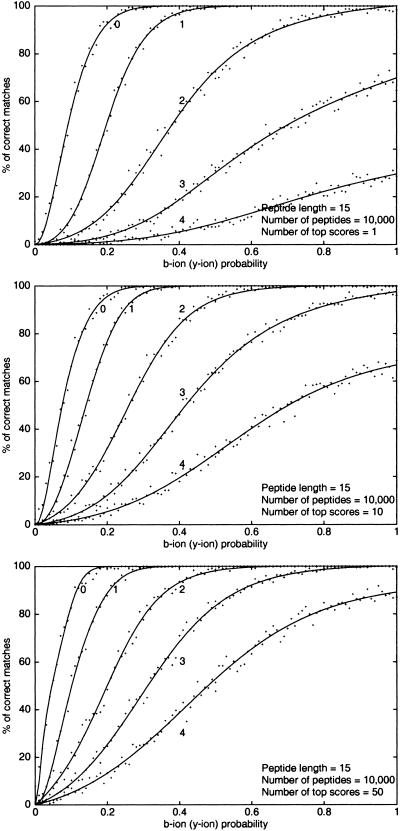
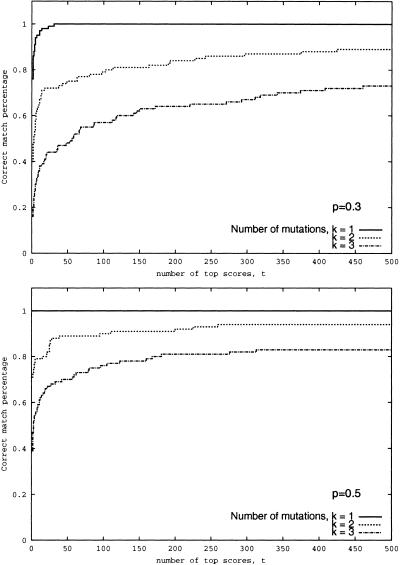
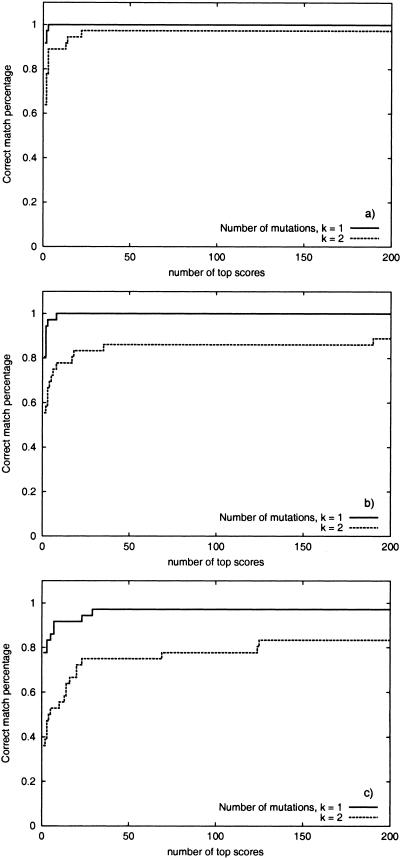
Although protein identification by matching tandem mass spectra (MS/MS) against protein databases is a widespread tool in mass spectrometry, the question about reliability of such searches remains open. Absence of rigorous significance scores in MS/MS database search makes it difficult to discard random database hits and may lead to erroneous protein identification, particularly in the case of mutated or post-translationally modified peptides. This problem is especially important for high-throughput MS/MS projects when the possibility of expert analysis is limited. Thus, algorithms that sort out reliable database hits from unreliable ones and identify mutated and modified peptides are sought. Most MS/MS database search algorithms rely on variations of the Shared Peaks Count approach that scores pairs of spectra by the peaks (masses) they have in common. Although this approach proved to be useful, it has a high error rate in identification of mutated and modified peptides. We describe new MS/MS database search tools, MS-CONVOLUTION and MS-ALIGNMENT, which implement the spectral convolution and spectral alignment approaches to peptide identification. We further analyze these approaches to identification of modified peptides and demonstrate their advantages over the Shared Peaks Count. We also use the spectral alignment approach as a filter in a new database search algorithm that reliably identifies peptides differing by up to two mutations/modifications from a peptide in a database.
Figures



Comment in
-
Measuring the dynamics of the proteome.Genome Res. 2001 Feb;11(2):191-3. doi: 10.1101/gr.178301. Genome Res. 2001. PMID: 11157781 No abstract available.
References
-
- Blom N, Gammeltoft S, Brunak S. Sequence- and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294:1351–1362. - PubMed
-
- Bushnell ML, Chen X. Efficient Branch and Bound Search With Application to Computer-Aided Design. Kluwer Academic Publishers; 1996.
-
- Clauser KR, Baker PR, Burlingame AL. The role of accurate mass measurement ( + /– 10ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal Chem. 1999;71:2871–2882. - PubMed
-
- Dancik V, Addona T, Clauser K, Vath J, Pevzner PA. De novo peptide sequencing via tandem mass spectrometry. J Comp Biol. 1999;6:327–342. - PubMed
-
- Eng J, McCormack A, Yates J. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Amer Soc Mass Spect. 1994;5:976–989. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources