

Defective repression of c-myc in breast cancer cells: A loss at the core of the transforming growth factor beta growth arrest program
- PMID: 11158583
- PMCID: PMC14697
- DOI: 10.1073/pnas.98.3.992
Defective repression of c-myc in breast cancer cells: A loss at the core of the transforming growth factor beta growth arrest program
Abstract
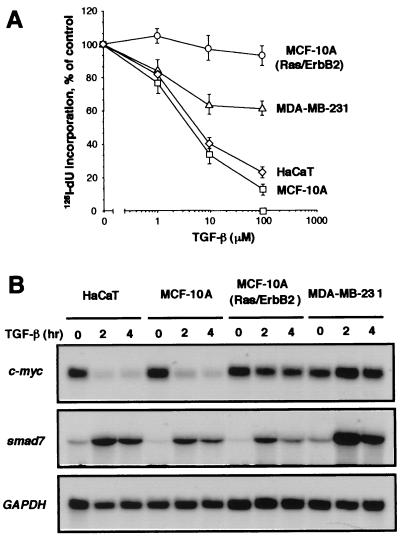
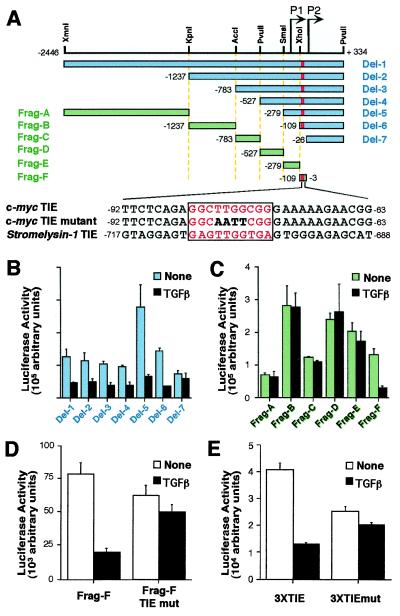
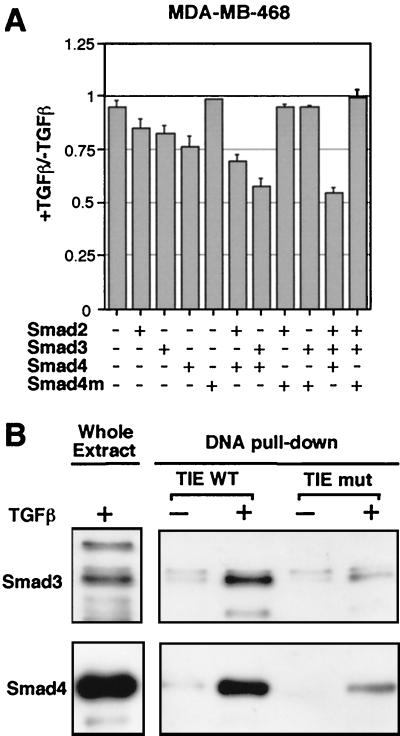
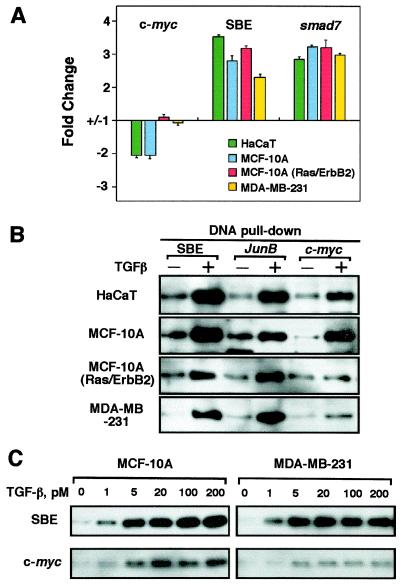
Loss of growth inhibitory responses to the cytokine transforming growth factor beta (TGF-beta) in cancer cells may result from mutational inactivation of TGF-beta receptors or their signal transducers, the Smad transcription factors. In breast cancer, however, loss of TGF-beta growth inhibition often occurs without a loss of these signaling components. A genome-wide analysis of rapid TGF-beta gene responses in MCF-10A human mammary epithelial cells and MDA-MB-231 breast cancer cells shows that c-myc repression, a response that is key to the TGF-beta program of cell cycle arrest, is selectively lost in the cancer cell line. Transformation of MCF-10A cells with c-Ha-ras and c-erbB2 oncogenes also led to a selective loss of c-myc repression and cell cycle arrest response. TGF-beta stimulation of epithelial cells rapidly induces the formation of a Smad complex that specifically recognizes a TGF-beta inhibitory element in the c-myc promoter. Formation of this complex is deficient in the oncogenically transformed breast cells. These results suggest that a Smad complex that specifically mediates c-myc repression is a target of oncogenic signals in breast cancer.
Figures
References
-
- Roberts A B, Sporn M B. In: Peptide Growth Factors and Their Receptors. Sporn M B, Roberts A B, editors. Heidelberg: Springer; 1990. pp. 419–472.
-
- Alexandrow M G, Moses H L. Cancer Res. 1995;55:1452–1457. - PubMed
-
- Massagué J. Nat Rev Mol Cell Biol. 2000;1:169–181. - PubMed
-
- Massagué J, Blain S W, Lo R S. Cell. 2000;103:295–309. - PubMed
-
- Derynck R, Zhang Y, Feng X H. Cell. 1998;95:737–740. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
