

Genetic remodeling of protein glycosylation in vivo induces autoimmune disease
- PMID: 11158608
- PMCID: PMC14722
- DOI: 10.1073/pnas.98.3.1142
Genetic remodeling of protein glycosylation in vivo induces autoimmune disease
Abstract
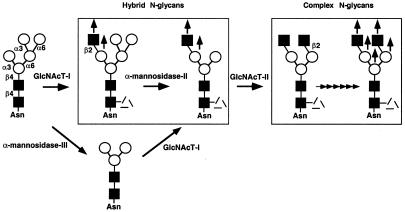
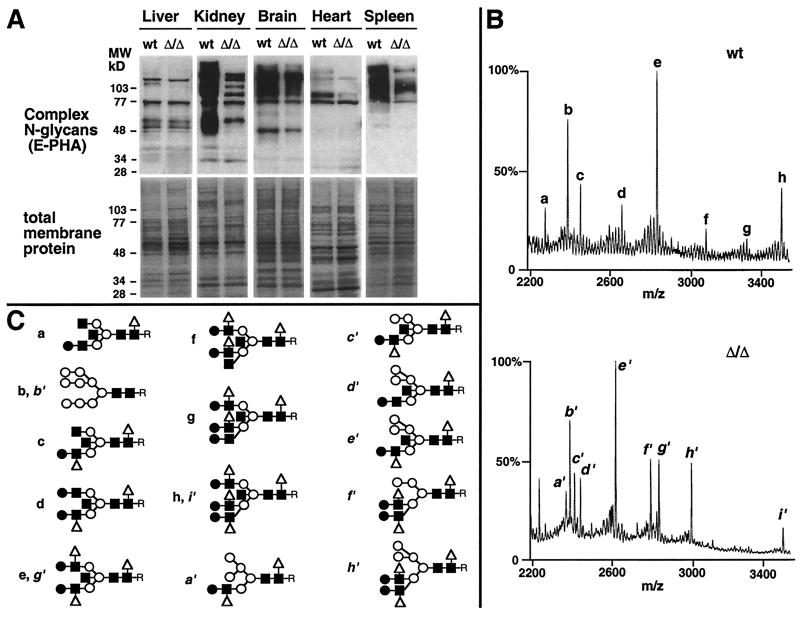
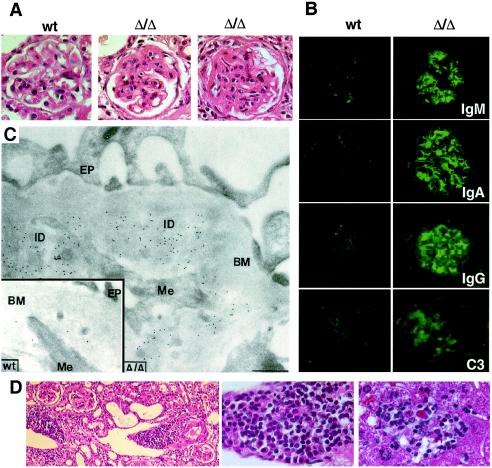
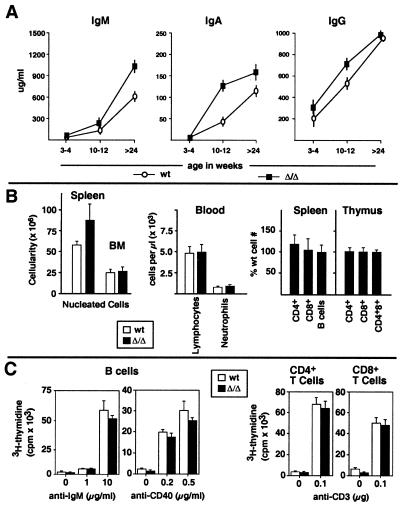
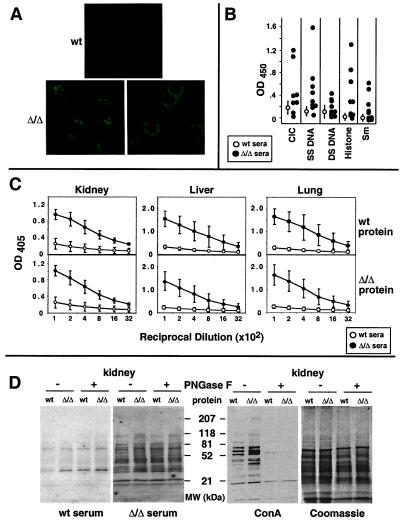
Autoimmune diseases are among the most prevalent of afflictions, yet the genetic factors responsible are largely undefined. Protein glycosylation in the Golgi apparatus produces structural variation at the cell surface and contributes to immune self-recognition. Altered protein glycosylation and antibodies that recognize endogenous glycans have been associated with various autoimmune syndromes, with the possibility that such abnormalities may reflect genetic defects in glycan formation. We show that mutation of a single gene, encoding alpha-mannosidase II, which regulates the hybrid to complex branching pattern of extracellular asparagine (N)-linked oligosaccharide chains (N-glycans), results in a systemic autoimmune disease similar to human systemic lupus erythematosus. alpha-Mannosidase II-deficient autoimmune disease is due to an incomplete overlap of two conjoined pathways in complex-type N-glycan production. Lymphocyte development, abundance, and activation parameters are normal; however, serum immunoglobulins are increased and kidney function progressively falters as a disorder consistent with lupus nephritis develops. Autoantibody reactivity and circulating immune complexes are induced, and anti-nuclear antibodies exhibit reactivity toward histone, Sm antigen, and DNA. These findings reveal a genetic cause of autoimmune disease provoked by a defect in the pathway of protein N-glycosylation.
Figures
References
-
- Steinman L. Cell. 1995;80:7–10. - PubMed
-
- Cohen P L. In: Fundamental Immunology. Paul W E, editor. Philadelphia: Lippincott; 1999. pp. 1067–1088.
-
- Gleeson P A. Biochim Biophys Acta. 1994;1197:237–255. - PubMed
-
- Delves P J. Autoimmunity. 1998;27:239–253. - PubMed
-
- Nyame A K, Debose-Boyd R, Long T D, Tsang V C W, Cummings R D. Glycobiology. 1998;8:615–624. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
