

Phylogenetic evidence for horizontal transfer of mutS alleles among naturally occurring Escherichia coli strains
- PMID: 11160094
- PMCID: PMC95048
- DOI: 10.1128/JB.183.5.1631-1644.2001
Phylogenetic evidence for horizontal transfer of mutS alleles among naturally occurring Escherichia coli strains
Abstract
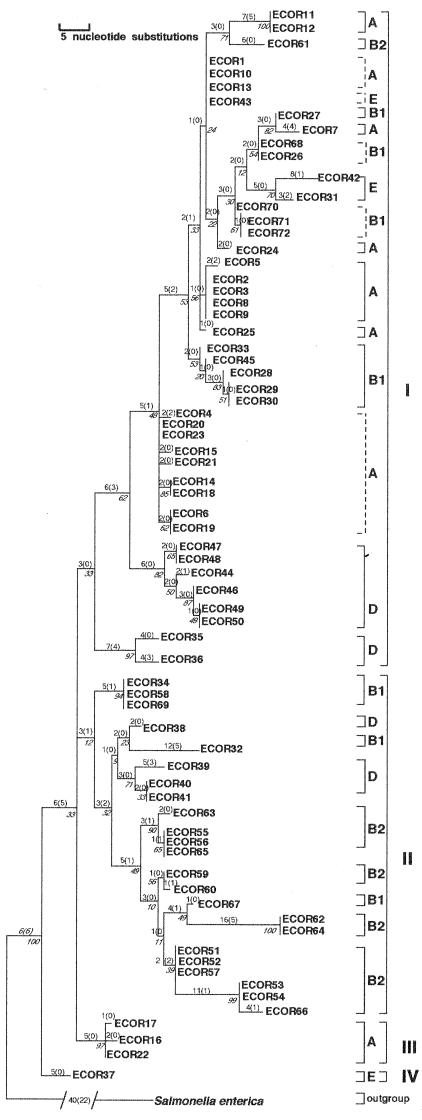
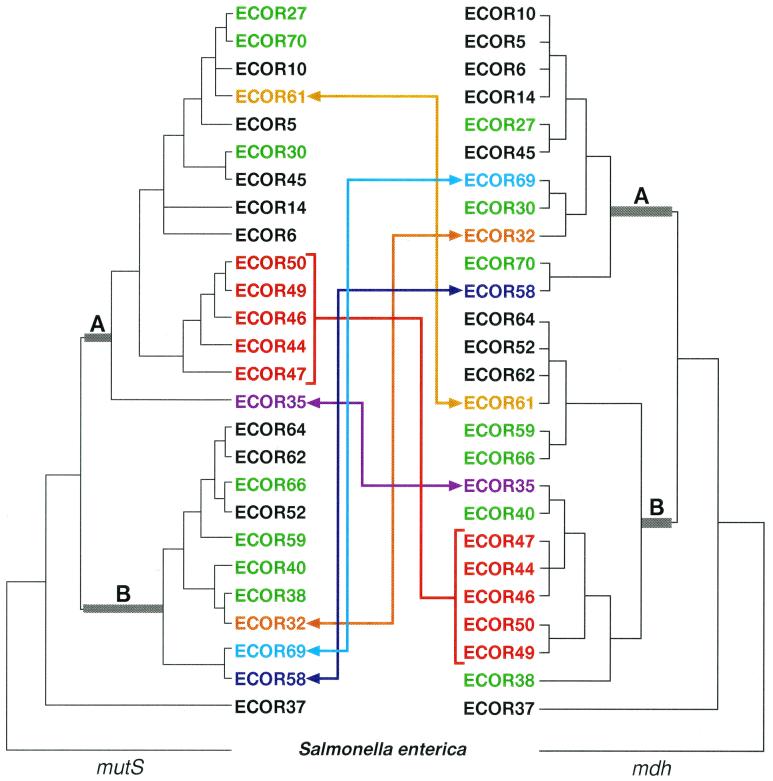
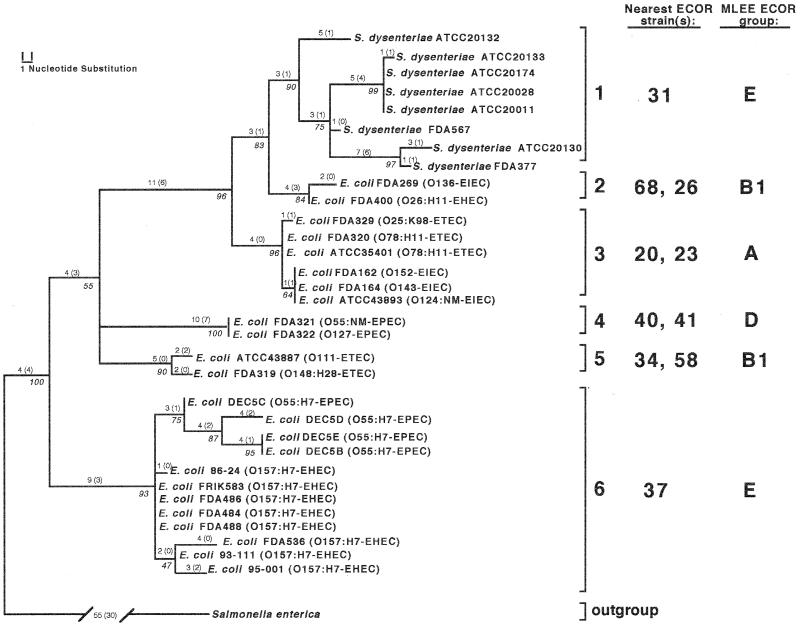
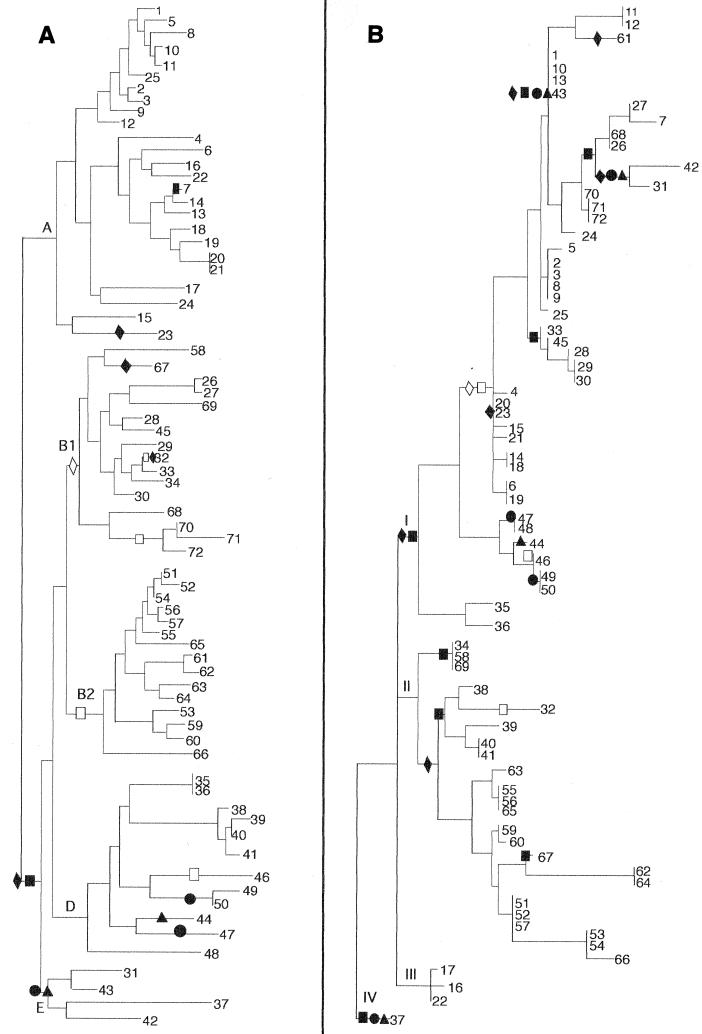
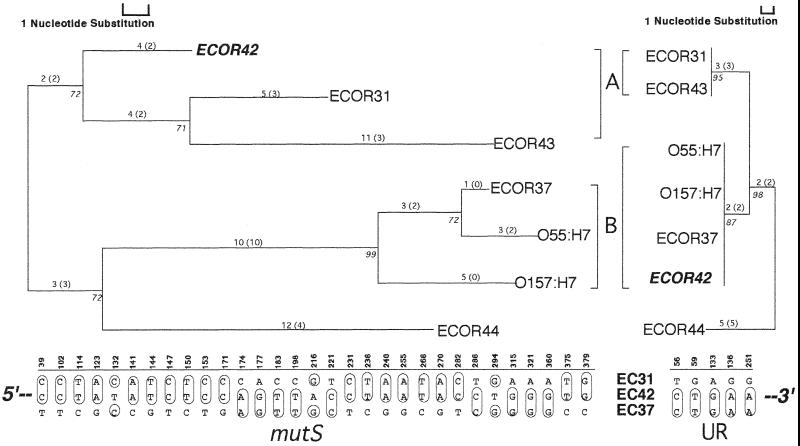
mutS mutators accelerate the bacterial mutation rate 100- to 1,000-fold and relax the barriers that normally restrict homeologous recombination. These mutators thus afford the opportunity for horizontal exchange of DNA between disparate strains. While much is known regarding the mutS phenotype, the evolutionary structure of the mutS(+) gene in Escherichia coli remains unclear. The physical proximity of mutS to an adjacent polymorphic region of the chromosome suggests that this gene itself may be subject to horizontal transfer and recombination events. To test this notion, a phylogenetic approach was employed that compared gene phylogeny to strain phylogeny, making it possible to identify E. coli strains in which mutS alleles have recombined. Comparison of mutS phylogeny against predicted E. coli "whole-chromosome" phylogenies (derived from multilocus enzyme electrophoresis and mdh sequences) revealed striking levels of phylogenetic discordance among mutS alleles and their respective strains. We interpret these incongruences as signatures of horizontal exchange among mutS alleles. Examination of additional sites surrounding mutS also revealed incongruous distributions compared to E. coli strain phylogeny. This suggests that other regional sequences are equally subject to horizontal transfer, supporting the hypothesis that the 61.5-min mutS-rpoS region is a recombinational hot spot within the E. coli chromosome. Furthermore, these data are consistent with a mechanism for stabilizing adaptive changes promoted by mutS mutators through rescue of defective mutS alleles with wild-type sequences.
Figures

References
-
- Bingen E, Picard B, Brahimi N, Mathy S, Desjardins P, Elion J, Denamur E. Phylogenetic analysis of Escherichia coli strains causing neonatal meningitis suggests horizontal gene transfer from a predominant pool of highly virulent B2 strains. J Infect Dis. 1998;177:642–50. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
