

Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism
- PMID: 11160820
- PMCID: PMC30565
- DOI: 10.1091/mbc.12.1.27
Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism
Abstract
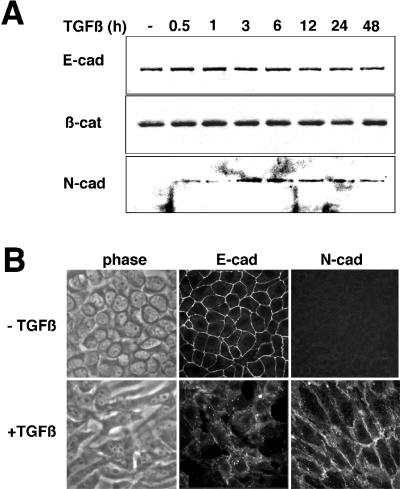
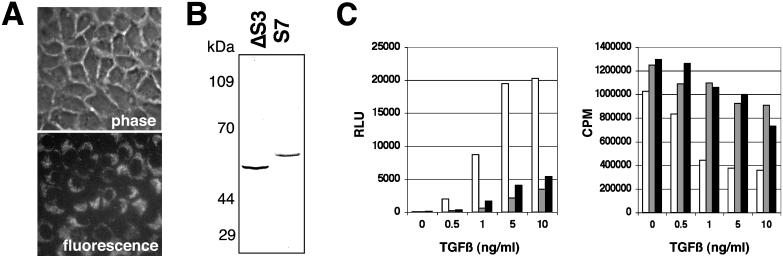
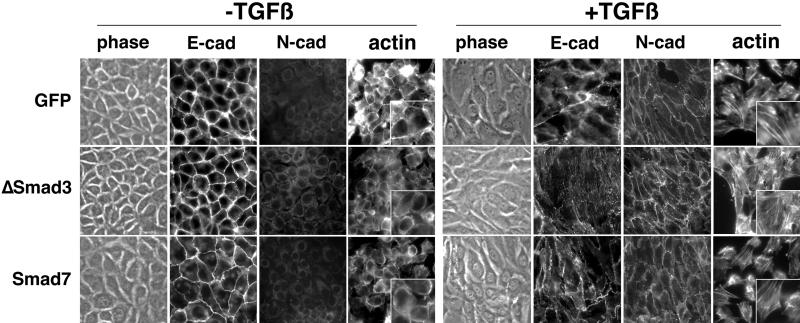
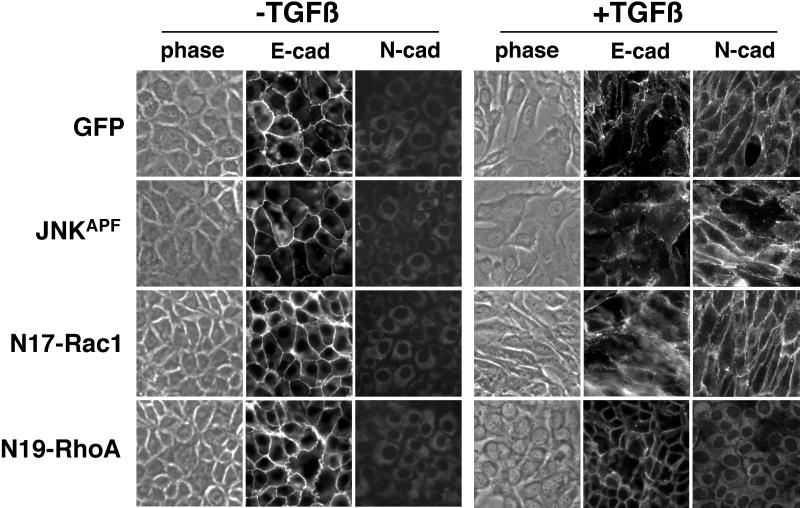
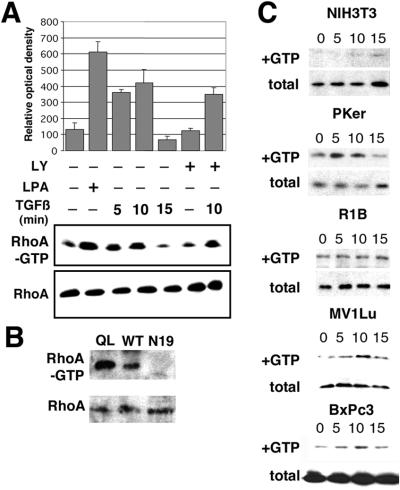
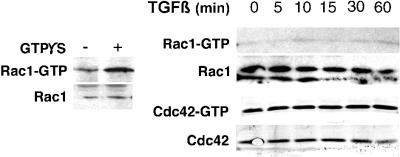
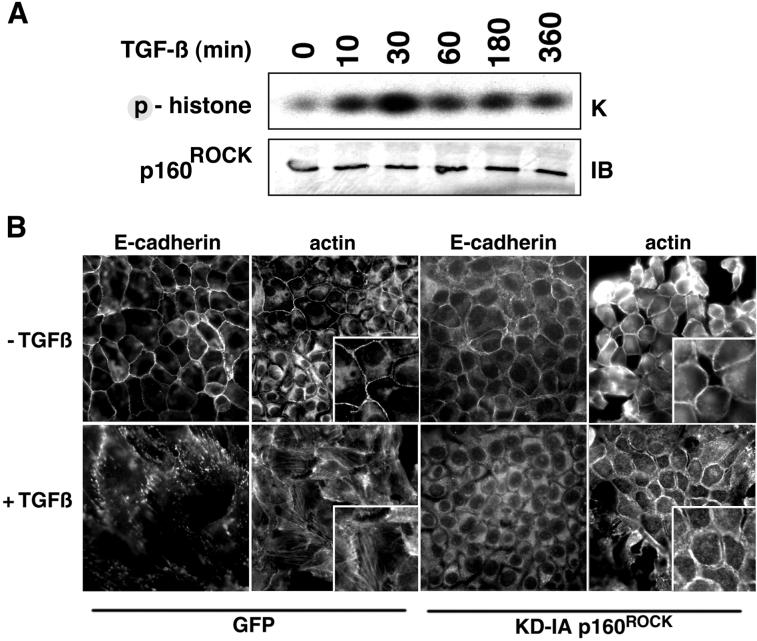
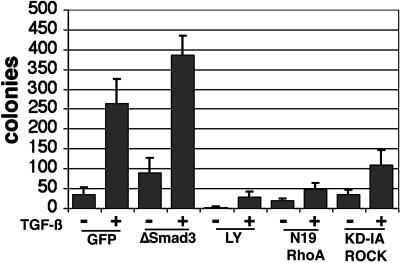
Transforming growth factor-beta1 (TGF-beta) can be tumor suppressive, but it can also enhance tumor progression by stimulating the complex process of epithelial-to-mesenchymal transdifferentiaion (EMT). The signaling pathway(s) that regulate EMT in response to TGF-beta are not well understood. We demonstrate the acquisition of a fibroblastoid morphology, increased N-cadherin expression, loss of junctional E-cadherin localization, and increased cellular motility as markers for TGF-beta-induced EMT. The expression of a dominant-negative Smad3 or the expression of Smad7 to levels that block growth inhibition and transcriptional responses to TGF-beta do not inhibit mesenchymal differentiation of mammary epithelial cells. In contrast, we show that TGF-beta rapidly activates RhoA in epithelial cells, and that blocking RhoA or its downstream target p160(ROCK), by the expression of dominant-negative mutants, inhibited TGF-beta-mediated EMT. The data suggest that TGF-beta rapidly activates RhoA-dependent signaling pathways to induce stress fiber formation and mesenchymal characteristics.
Figures
References
-
- Ashcroft GS, Yang X, Glick AB, Weinstein M, Letterio JL, Mizel DE, Anzano M, Greenwell-Wild T, Wahl SM, Deng C, Roberts AB. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol. 1999;1:260–266. - PubMed
-
- Atfi A, Djelloul S, Chastre E, Davis R, Gespach C. Evidence for a role of Rho-like GTPases and stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) in transforming growth factor β-mediated signaling. J Biol Chem. 1997;272:1429–1432. - PubMed
-
- Azuma M, Tamatani T, Fukui K, Yuki T, Motegi K, Sato M. Different signaling pathways involved in transforming growth factor-β1-induced morphological change and type IV collagen synthesis in simian virus-40-immortalized normal human salivary gland duct and myoepithelial cell clones. Arch Oral Biol. 1996;41:413–424. - PubMed
-
- Benard V, Bohl BP, Bokoch GM. Characterization of rac and cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J Biol Chem. 1999;274:13198–13204. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
