

Digging for dead genes: an analysis of the characteristics of the pseudogene population in the Caenorhabditis elegans genome
- PMID: 11160906
- PMCID: PMC30377
- DOI: 10.1093/nar/29.3.818
Digging for dead genes: an analysis of the characteristics of the pseudogene population in the Caenorhabditis elegans genome
Abstract
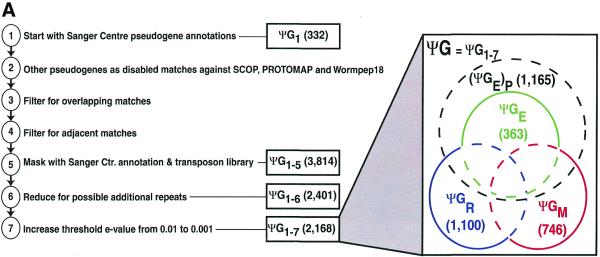
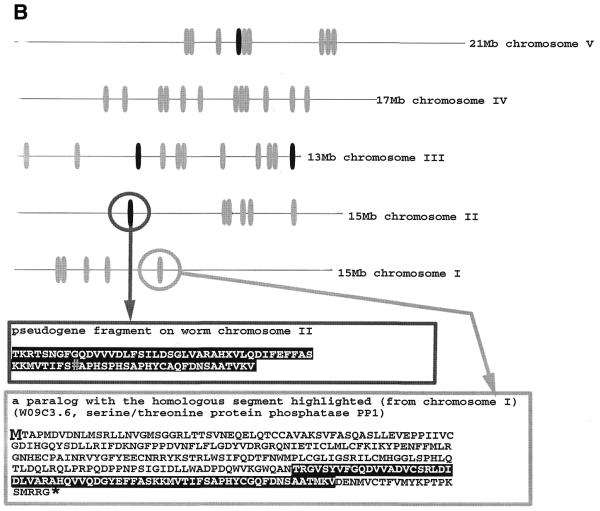
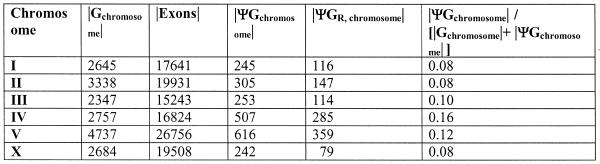
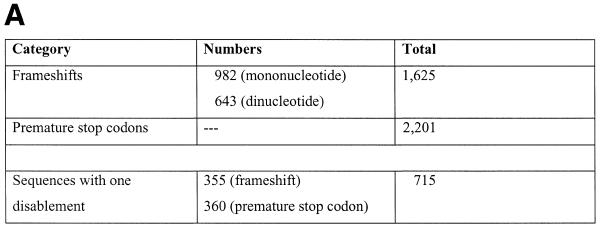
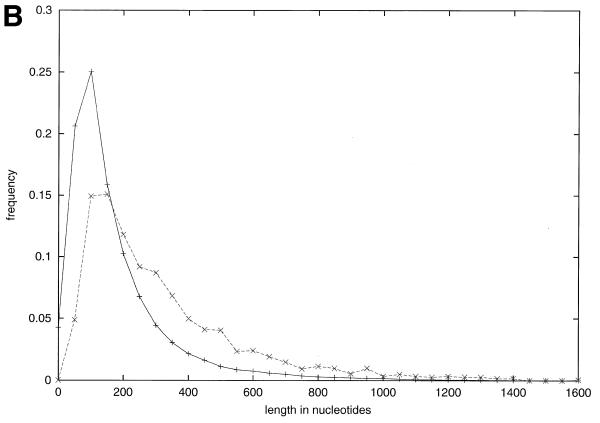
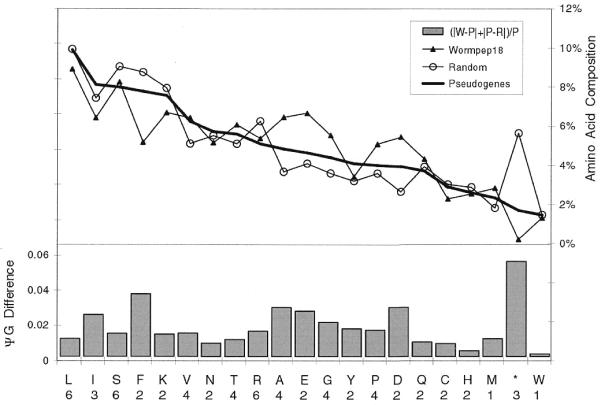
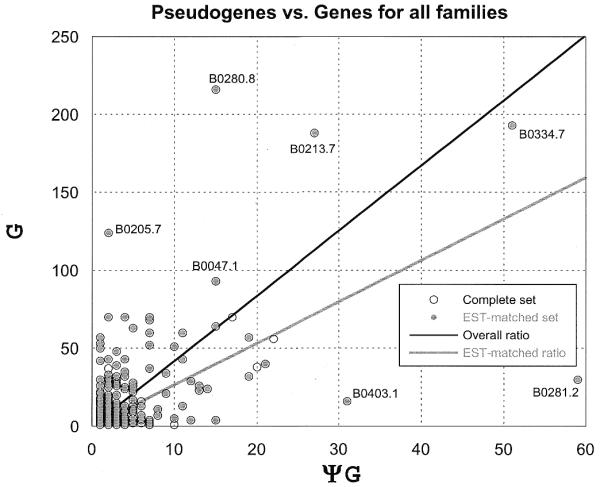
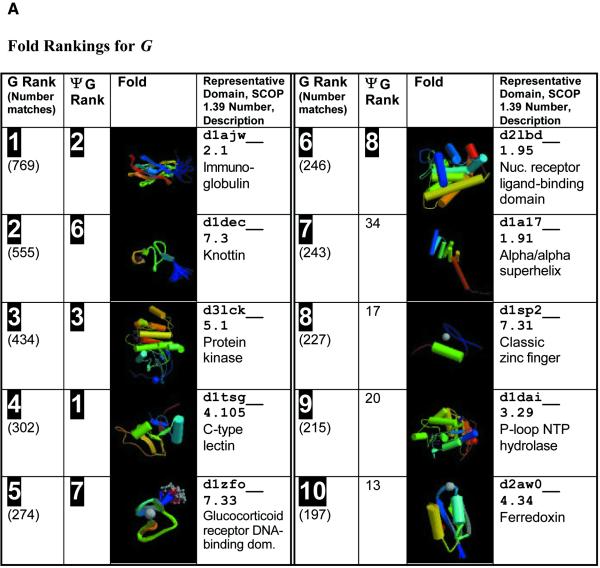
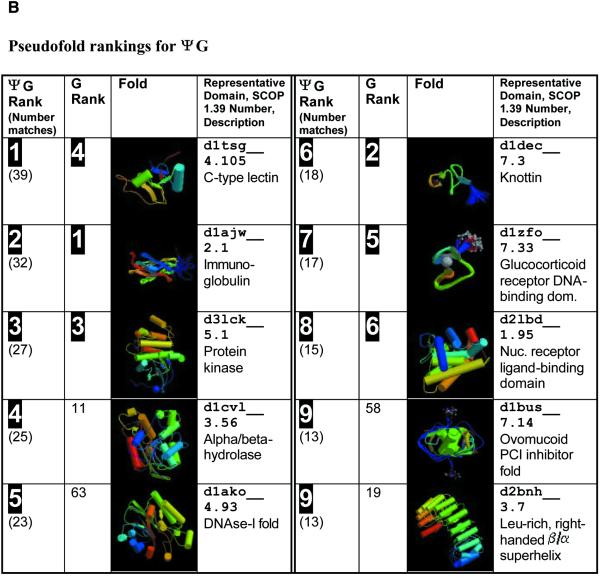
Pseudogenes are non-functioning copies of genes in genomic DNA, which may either result from reverse transcription from an mRNA transcript (processed pseudogenes) or from gene duplication and subsequent disablement (non-processed pseudogenes). As pseudogenes are apparently 'dead', they usually have a variety of obvious disablements (e.g., insertions, deletions, frameshifts and truncations) relative to their functioning homologs. We have derived an initial estimate of the size, distribution and characteristics of the pseudogene population in the Caenorhabditis elegans genome, performing a survey in 'molecular archaeology'. Corresponding to the 18 576 annotated proteins in the worm (i.e., in Wormpep18), we have found an estimated total of 2168 pseudogenes, about one for every eight genes. Few of these appear to be processed. Details of our pseudogene assignments are available from http://bioinfo.mbb.yale.edu/genome/worm/pseudogene. The population of pseudogenes differs significantly from that of genes in a number of respects: (i) pseudogenes are distributed unevenly across the genome relative to genes, with a disproportionate number on chromosome IV; (ii) the density of pseudogenes is higher on the arms of the chromosomes; (iii) the amino acid composition of pseudogenes is midway between that of genes and (translations of) random intergenic DNA, with enrichment of Phe, Ile, Leu and Lys, and depletion of Asp, Ala, Glu and Gly relative to the worm proteome; and (iv) the most common protein folds and families differ somewhat between genes and pseudogenes-whereas the most common fold found in the worm proteome is the immunoglobulin fold and the most common 'pseudofold' is the C-type lectin. In addition, the size of a gene family bears little overall relationship to the size of its corresponding pseudogene complement, indicating a highly dynamic genome. There are in fact a number of families associated with large populations of pseudogenes. For example, one family of seven-transmembrane receptors (represented by gene B0334.7) has one pseudogene for every four genes, and another uncharacterized family (represented by gene B0403.1) is approximately two-thirds pseudogenic. Furthermore, over a hundred apparent pseudogenic fragments do not have any obvious homologs in the worm.
Figures

References
-
- Weiner A.M., Deininger,P.L. and Efstratiadis,A. (1986) Non-viral retroposons: genes, pseudogenes and transposable elements generated by the reverse flow of genetic information. Annu. Rev. Biochem., 55, 631–661. - PubMed
-
- Vanin E.F. (1985) Processed pseudogenes: characteristics and evolution. Annu. Rev. Genet., 19, 253–272. - PubMed
-
- Mighell A.J., Smith,N.R., Robinson,P.A. and Markham,A.F. (2000) Vertebrate pseudogenes. FEBS Lett., 468, 109–114. - PubMed
-
- Olsen M.A. and Schechter,L.E. (1999) Cloning, mRNA localization and evolutionary conservation of a human 5HT7 receptor pseudogene. Gene, 227, 63–69. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
