

The molecular basis of 3-methylcrotonylglycinuria, a disorder of leucine catabolism
- PMID: 11170888
- PMCID: PMC1235267
- DOI: 10.1086/318202
The molecular basis of 3-methylcrotonylglycinuria, a disorder of leucine catabolism
Abstract
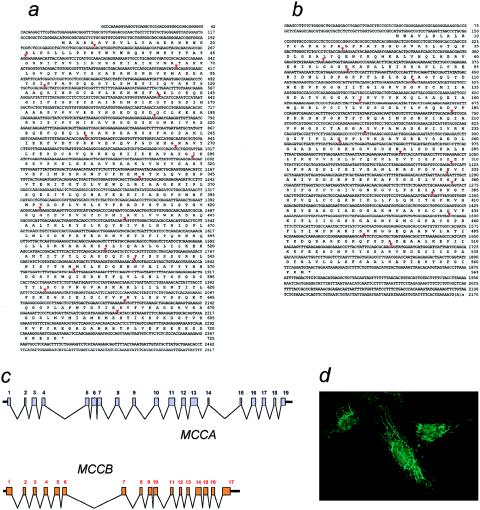
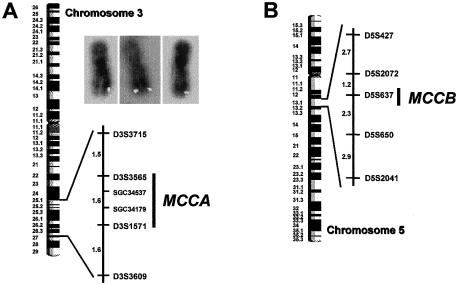
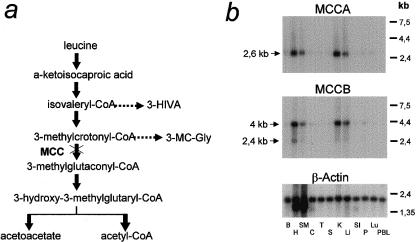
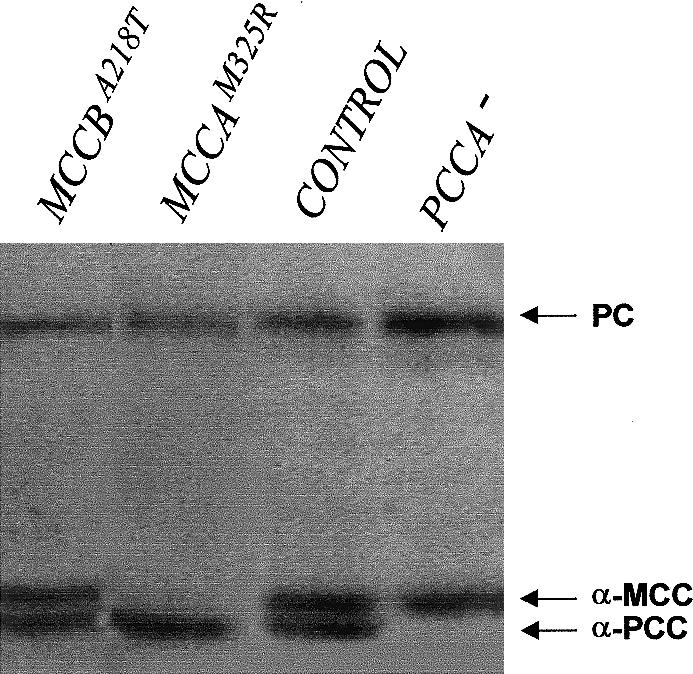
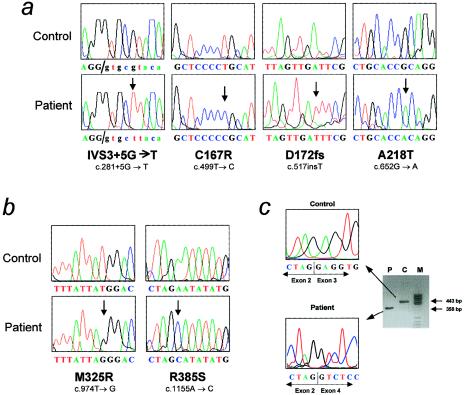
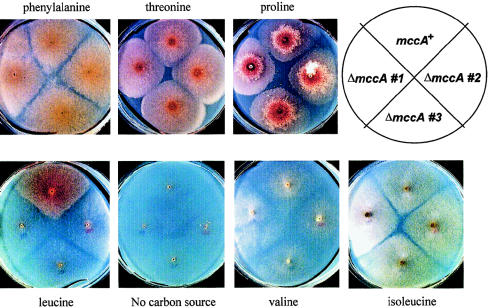
3-Methylcrotonylglycinuria is an inborn error of leucine catabolism and has a recessive pattern of inheritance that results from the deficiency of 3-methylcrotonyl-CoA carboxylase (MCC). The introduction of tandem mass spectrometry in newborn screening has revealed an unexpectedly high incidence of this disorder, which, in certain areas, appears to be the most frequent organic aciduria. MCC, an heteromeric enzyme consisting of alpha (biotin-containing) and beta subunits, is the only one of the four biotin-dependent carboxylases known in humans that has genes that have not yet been characterized, precluding molecular studies of this disease. Here we report the characterization, at the genomic level and at the cDNA level, of both the MCCA gene and the MCCB gene, encoding the MCC alpha and MCC beta subunits, respectively. The 19-exon MCCA gene maps to 3q25-27 and encodes a 725-residue protein with a biotin attachment site; the 17-exon MCCB gene maps to 5q12-q13 and encodes a 563-residue polypeptide. We show that disease-causing mutations can be classified into two complementation groups, denoted "CGA" and "CGB." We detected two MCCA missense mutations in CGA patients, one of which leads to absence of biotinylated MCC alpha. Two MCCB missense mutations and one splicing defect mutation leading to early MCC beta truncation were found in CGB patients. A fourth MCCB mutation also leading to early MCC beta truncation was found in two nonclassified patients. A fungal model carrying an mccA null allele has been constructed and was used to demonstrate, in vivo, the involvement of MCC in leucine catabolism. These results establish that 3-methylcrotonylglycinuria results from loss-of-function mutations in the genes encoding the alpha and beta subunits of MCC and complete the genetic characterization of the four human biotin-dependent carboxylases.
Figures
References
Electronic-Database Information
-
- Expressed Sequence Tags database, http://www.ncbi.nlm.nih.gov/dbEST/index.html
-
- Fungal (Aspergillus nidulans, Aspergillus parasiticus, and Neurospora crassa) Cosmid and cDNA Sequencing, http://www.genome.ou.edu/fungal.html (for Aspergillus nidulans [accession number n1c10])
-
- Fungal Genetics Stock Center, http://www.fgsc.net/ (for fungal EST clones)
-
- GenBank Overview, http://www.ncbi.nlm.nih.gov/Genbank/GenbankOverview.html (for human MCCA ESTs, [accession numbers AA134548, AA605162, and AW410916], human MCCB ESTs [accession numbers AA465612, AI949422, AI367183, AW439494, and AI869038], full-length human MCCA cDNA [accession number AF310972], full-length MCCB cDNA [accession number AF310971], human MCCA BAC [accession number AC026920], and human MCCB BACs [accession numbers AC010279, AC026775, and AC025958])
References
-
- Beltrán-Valero de Bernabé D, Granadino B, Chiarelli I, Porfirio B, Mayatepek E, Aquaron R, Moore MM, Festen JJ, Sanmartí R, Peñalva MA, Rodríguez de Córdoba S (1998) Mutation and polymorphism analysis of the human homogentisate 1, 2- dioxygenase gene in alkaptonuria patients. Am J Hum Genet 62:776–784 - PMC - PubMed
-
- Chuang DT, Shih VE (1995) Disorders of branched chain amino acid and ketoacid metabolism. In: Scriver C, Beaudet A, Sly W, Valle D (eds) The metabolic basis of inherited disease, 7th ed. McGraw-Hill, New York, pp 1239–1276
-
- Cotton RGH, Horaitis O (2000) Quality control in the discovery, reporting and recording of genomic variation. Hum Mutat 15:16–21 - PubMed
-
- Fernández-Cañón JM, Granadino B, Beltrán-Valero de Bernabé D, Renedo M, Fernández-Ruiz E, Peñalva MA, Rodríguez de Córdoba S (1996) The molecular basis of alkaptonuria. Nat Genet 14:19–24 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
