

Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis
- PMID: 11179007
- PMCID: PMC1274472
- DOI: 10.1086/318796
Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis
Abstract
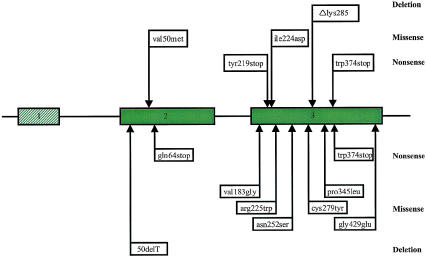
Familial hemophagocytic lymphohistiocytosis (FHL) is an autosomal recessive disease of early childhood characterized by nonmalignant accumulation and multivisceral infiltration of activated T lymphocytes and histiocytes (macrophages). Cytotoxic T and natural killer (NK) cell activity is markedly reduced or absent in these patients, and mutations in a lytic granule constituent, perforin, were recently identified in a number of FHL individuals. Here, we report a comprehensive survey of 34 additional patients with FHL for mutations in the coding region of the perforin gene and the relative frequency of perforin mutations in FHL. Perforin mutations were identified in 7 of the 34 families investigated. Six children were homozygous for the mutations, and one patient was a compound heterozygote. Four novel mutations were detected: one nonsense, two missense, and one deletion of one amino acid. In four families, a previously reported mutation at codon 374, causing a premature stop codon, was identified, and, therefore, this is the most common perforin mutation identified so far in FHL patients. We found perforin mutations in 20% of all FHL patients investigated (7/34), with a somewhat higher prevalence, approximately 30% (6/20), in children whose parents originated from Turkey. No other correlation between the type of mutation and the phenotype of the patients was evident from the present study. Our combined results from mutational analysis of 34 families and linkage analysis of a subset of consanguineous families indicate that perforin mutations account for 20%-40% of the FHL cases and the FHL 1 locus on chromosome 9 for approximately 10%, whereas the major part of the FHL cases are caused by mutations in not-yet-identified genes.
Figures
References
Electronic-Database Information
-
- Genbank, http://www.ncbi.nlm.nih.gov/Genbank (accession number for perforin [NM-005041] and [M28393])
-
- National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/genemap98 (for Genemap'98)
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for familial hemophagocytic lymphohistiocytosis [MIM 267700], familial hemophagocytic lymphohistiocytosis 1 [MIM 603552], familial hemophagocytic lymphohistiocytosis 2 [MIM 603553], Chediak-Higashi syndrome [MIM 214500], X-linked lymphoproliferative disease [MIM 308240], and Griscelli syndrome [MIM 214450])
References
-
- Darmon AJ, Bleackley RC (1998) Proteases and cell-mediated cytotoxicity. Crit Rev Immunol 18:255–273 - PubMed
-
- Darmon AJ, Nicholson DW, Bleackley RC (1995) Activation of the apoptotic protease CPP32 by cytotoxic T-cell-derived granzyme B. Nature 377:446–448 - PubMed
-
- Dufourcq-Lagelouse R, Pastural E, Barrat FJ, Feldmann J, Le Deist F, Fischer A, de Saint Basile G (1999b) Genetic basis of hemophagocytic lymphohistiocytosis syndrome. Int J Mol Med 4:127–133 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
