

Control of mitochondrial membrane permeabilization by adenine nucleotide translocator interacting with HIV-1 viral protein rR and Bcl-2
- PMID: 11181702
- PMCID: PMC2195906
- DOI: 10.1084/jem.193.4.509
Control of mitochondrial membrane permeabilization by adenine nucleotide translocator interacting with HIV-1 viral protein rR and Bcl-2
Abstract
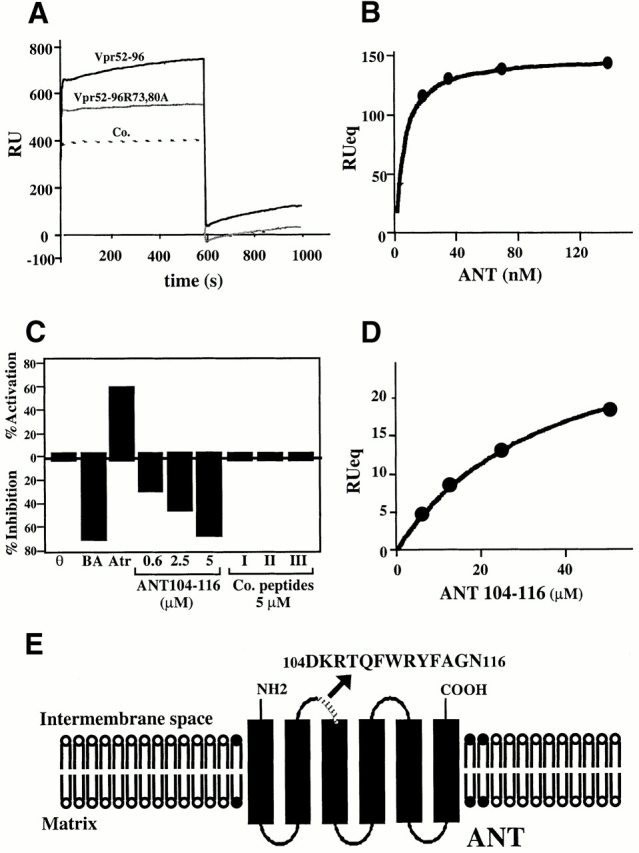
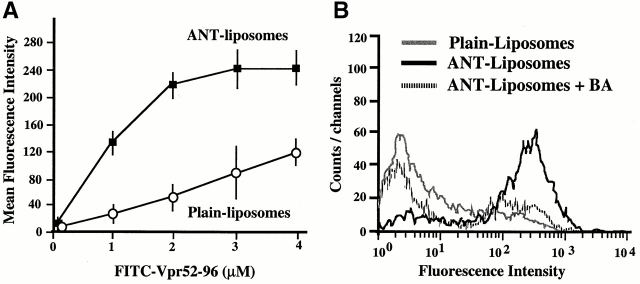
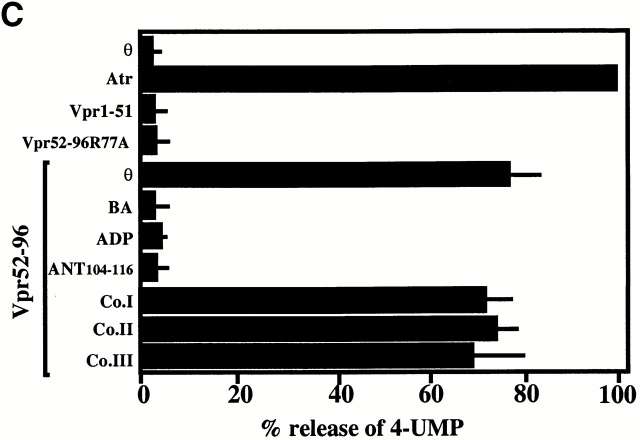
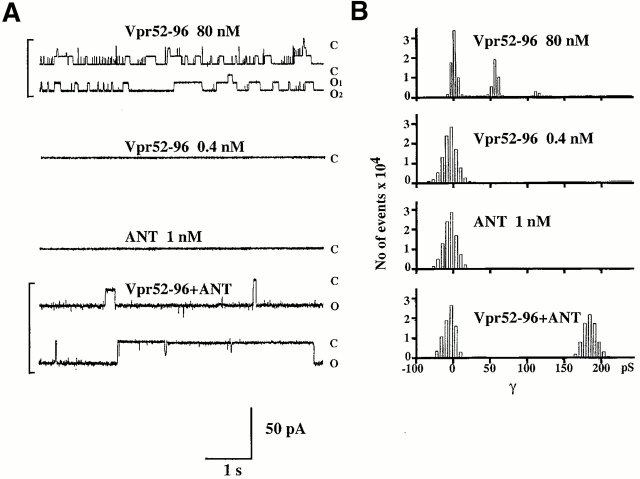
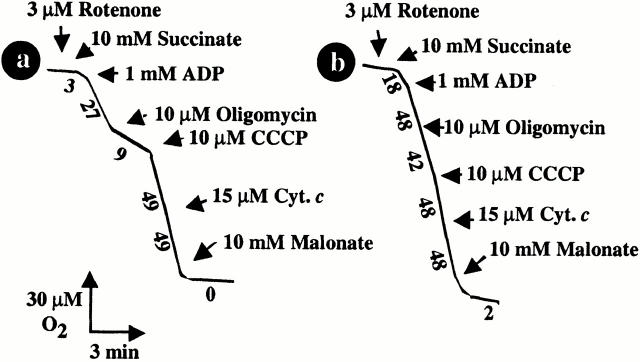
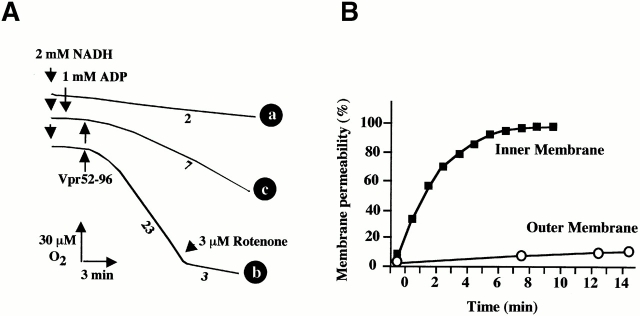
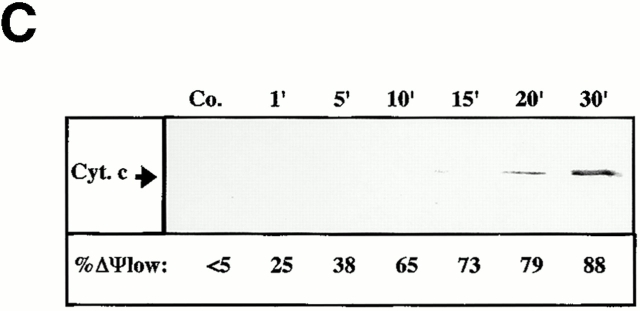
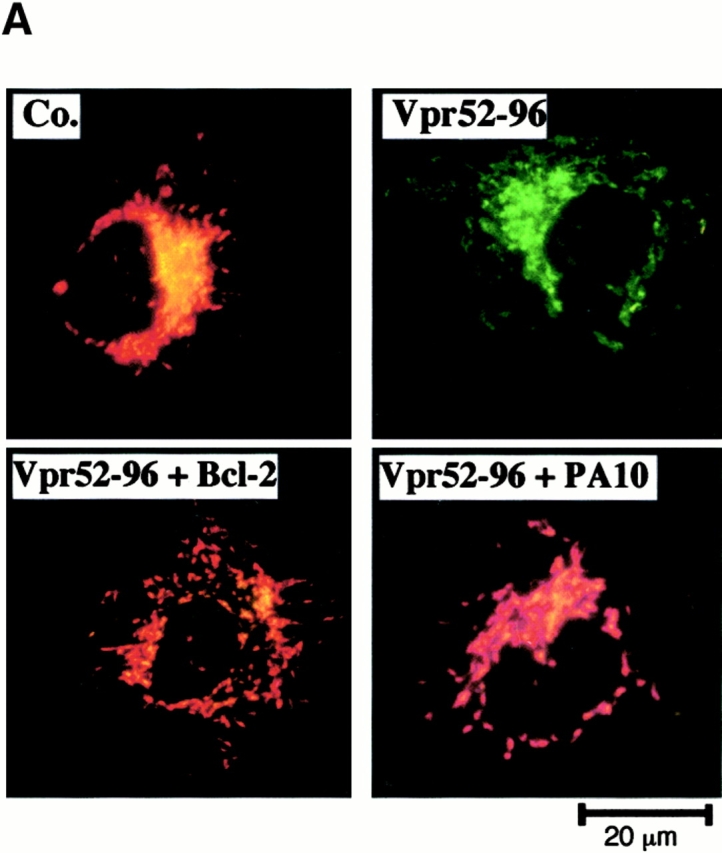
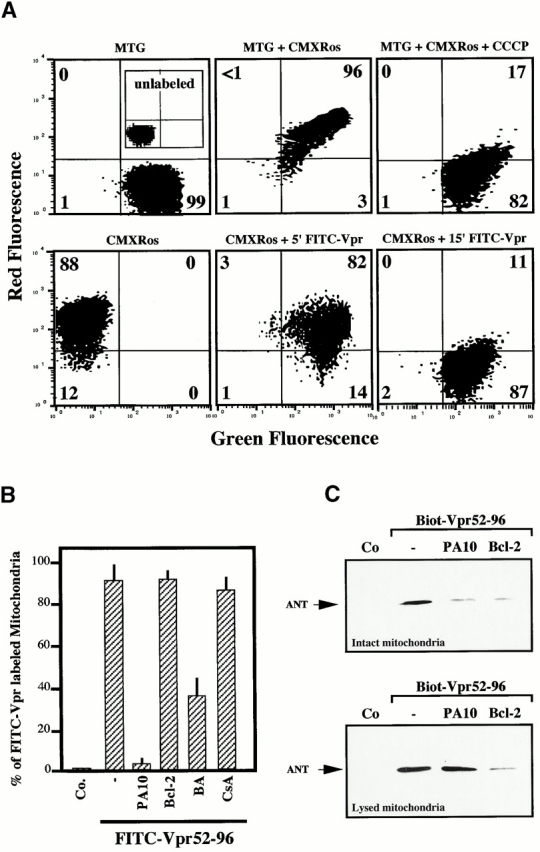
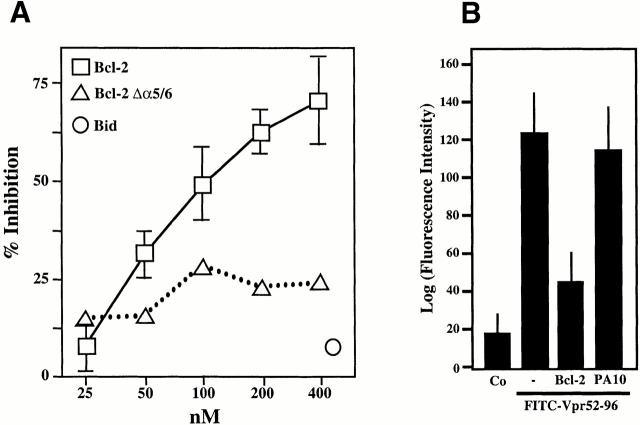
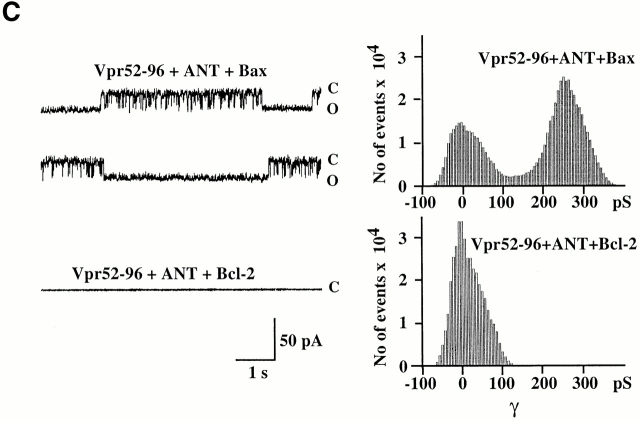
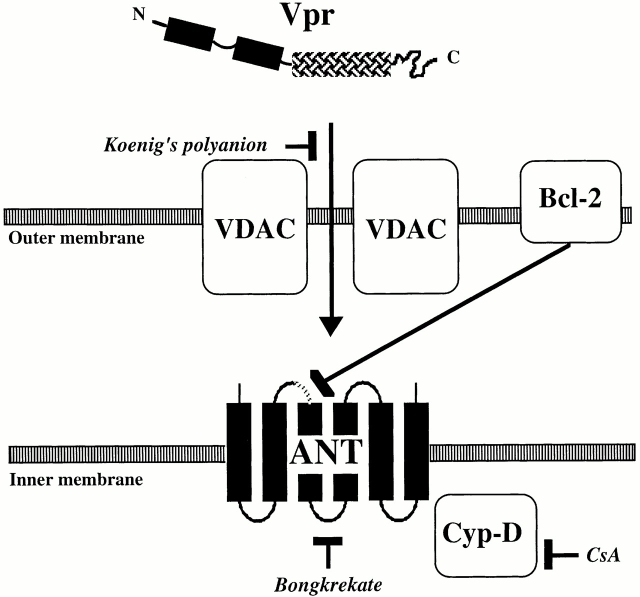
Viral protein R (Vpr), an apoptogenic accessory protein encoded by HIV-1, induces mitochondrial membrane permeabilization (MMP) via a specific interaction with the permeability transition pore complex, which comprises the voltage-dependent anion channel (VDAC) in the outer membrane (OM) and the adenine nucleotide translocator (ANT) in the inner membrane. Here, we demonstrate that a synthetic Vpr-derived peptide (Vpr52-96) specifically binds to the intermembrane face of the ANT with an affinity in the nanomolar range. Taking advantage of this specific interaction, we determined the role of ANT in the control of MMP. In planar lipid bilayers, Vpr52-96 and purified ANT cooperatively form large conductance channels. This cooperative channel formation relies on a direct protein-protein interaction since it is abolished by the addition of a peptide corresponding to the Vpr binding site of ANT. When added to isolated mitochondria, Vpr52-96 uncouples the respiratory chain and induces a rapid inner MMP to protons and NADH. This inner MMP precedes outer MMP to cytochrome c. Vpr52-96-induced matrix swelling and inner MMP both are prevented by preincubation of purified mitochondria with recombinant Bcl-2 protein. In contrast to König's polyanion (PA10), a specific inhibitor of the VDAC, Bcl-2 fails to prevent Vpr52-96 from crossing the mitochondrial OM. Rather, Bcl-2 reduces the ANT-Vpr interaction, as determined by affinity purification and plasmon resonance studies. Concomitantly, Bcl-2 suppresses channel formation by the ANT-Vpr complex in synthetic membranes. In conclusion, both Vpr and Bcl-2 modulate MMP through a direct interaction with ANT.
Figures
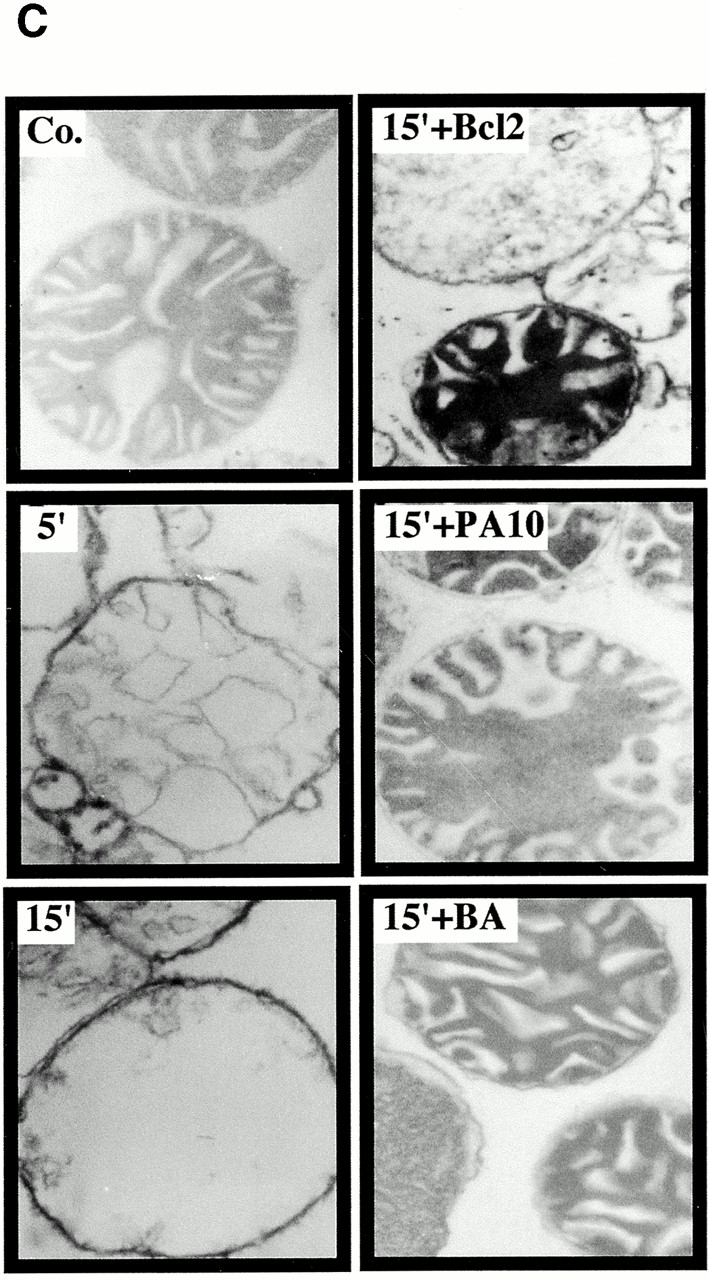
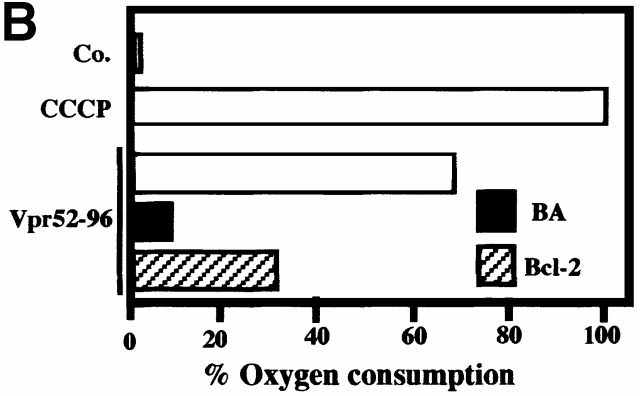
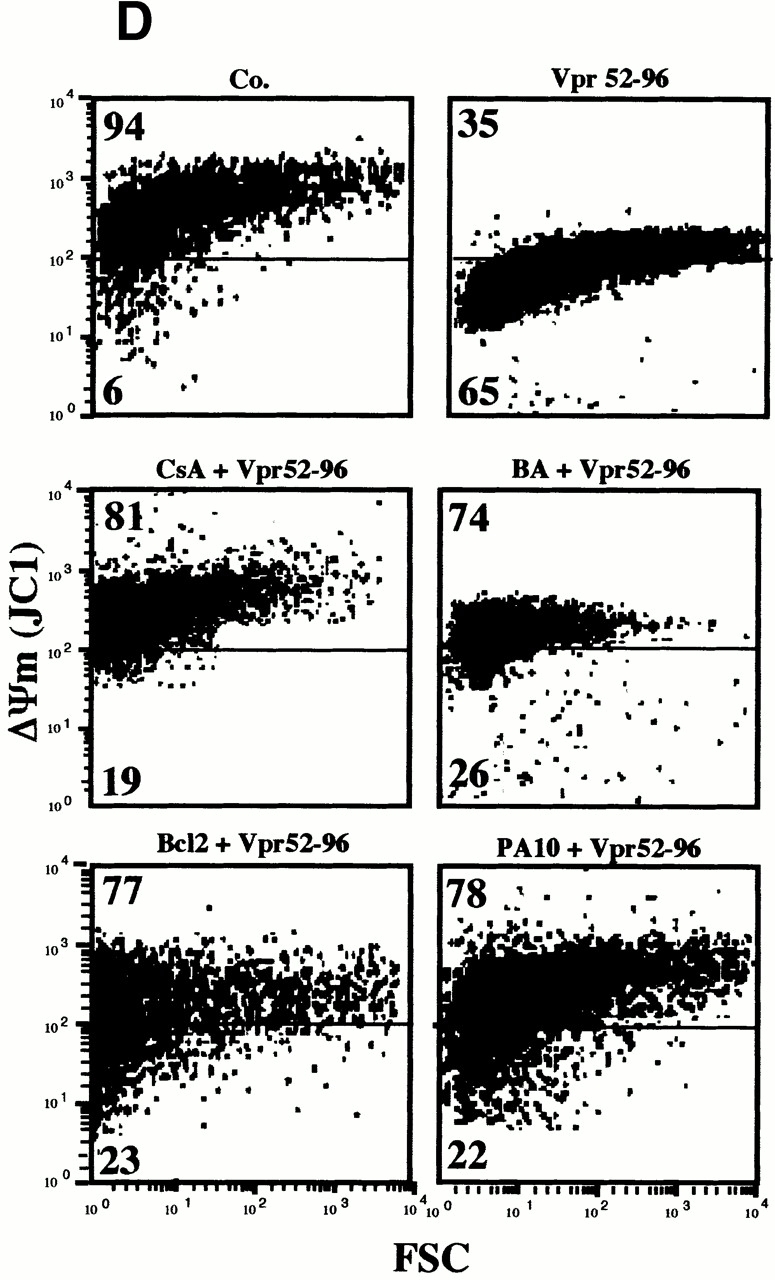
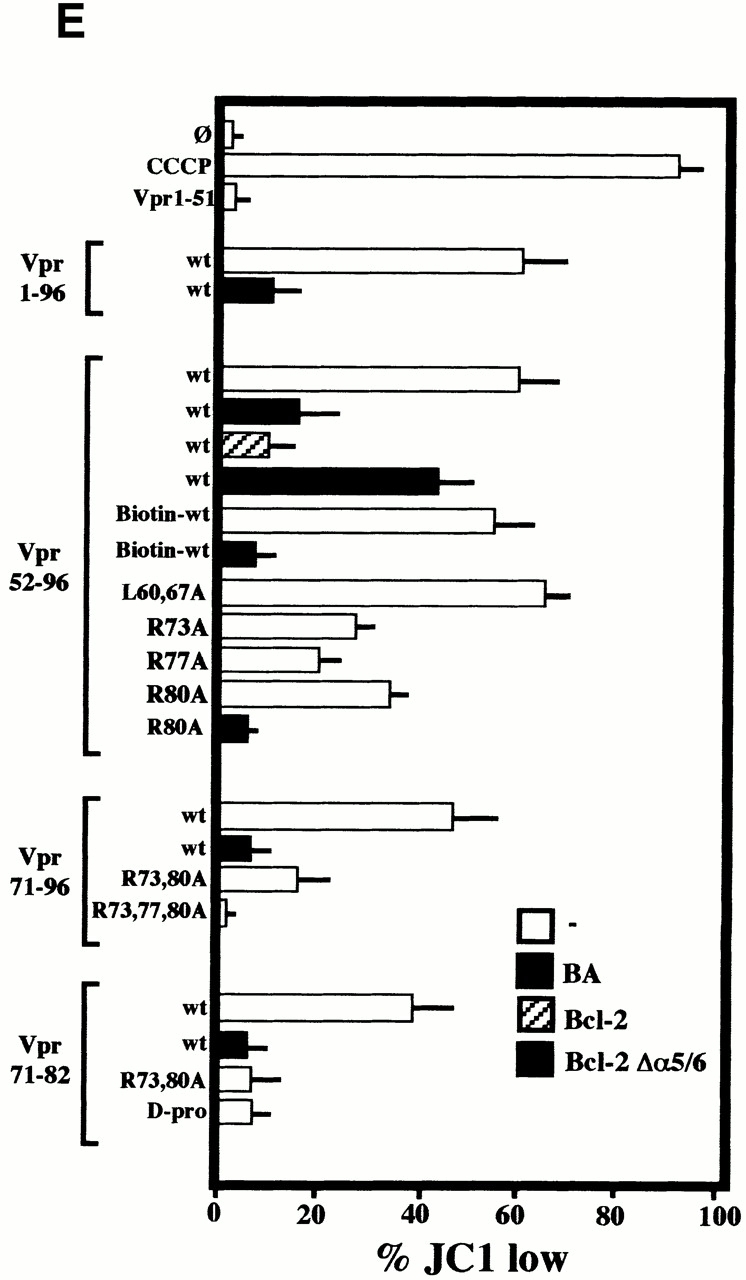
Comment in
-
HIV and apoptosis death and the mitochondrion.J Exp Med. 2001 Feb 19;193(4):F11-4. doi: 10.1084/jem.193.4.f11. J Exp Med. 2001. PMID: 11181706 Free PMC article. No abstract available.
References
-
- Kroemer G., Zamzami N., Susin S.A. Mitochondrial control of apoptosis. Immunol. Today. 1997;18:44–51. - PubMed
-
- Green D.R., Reed J.C. Mitochondria and apoptosis. Science. 1998;281:1309–1312. - PubMed
-
- Lemasters J.J., Nieminen A.-L., Qjan T., Trost L.C., Elmore S.P., Nishimura Y., Crowe R.A., Cascio W.E., Bradham C.A., Brenner D.A., Herman B. The mitochondrial permeability transition in cell deatha common mechanism in necrosis, apoptosis and autophagy. Biochim. Biophys. Acta. 1998;1366:177–196. - PubMed
-
- Wallace D.C. Mitochondrial diseases in mouse and man. Science. 1999;283:1482–1488. - PubMed
-
- Vander Heiden M.G., Thompson C.B. Bcl-2 proteinsinhibitors of apoptosis or regulators of mitochondrial homeostasis? Nat. Cell Biol. 1999;1:E209–E216. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
