

Point mutations in the murine fumarylacetoacetate hydrolase gene: Animal models for the human genetic disorder hereditary tyrosinemia type 1
- PMID: 11209059
- PMCID: PMC14641
- DOI: 10.1073/pnas.98.2.641
Point mutations in the murine fumarylacetoacetate hydrolase gene: Animal models for the human genetic disorder hereditary tyrosinemia type 1
Abstract
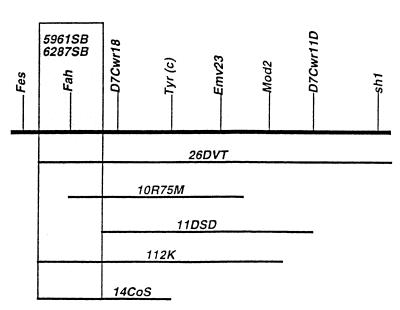
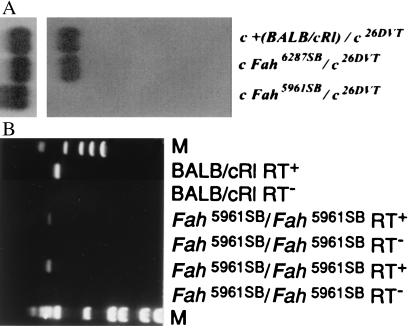
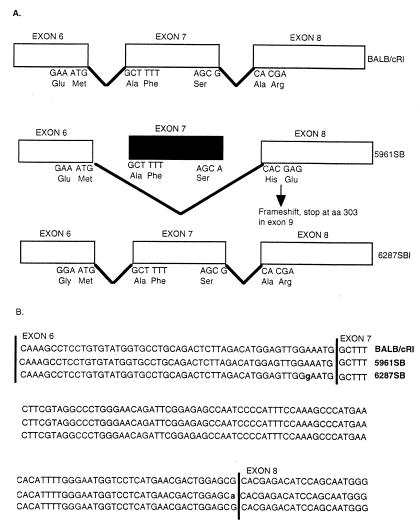
Hereditary tyrosinemia type 1 (HT1) is a severe autosomal recessive metabolic disease associated with point mutations in the human fumarylacetoacetate hydrolase (FAH) gene that disrupt tyrosine catabolism. An acute form of HT1 results in death during the first months of life because of hepatic failure, whereas a chronic form leads to gradual development of liver disease often accompanied by renal dysfunction, childhood rickets, neurological crisis, and hepatocellular carcinoma. Mice homozygous for certain chromosome 7 deletions of the albino Tyr; c locus that also include Fah die perinatally as a result of liver dysfunction and exhibit a complex syndrome characterized by structural abnormalities and alterations in gene expression in the liver and kidney. Here we report that two independent, postnatally lethal mutations induced by N-ethyl-N-nitrosourea and mapped near Tyr are alleles of Fah. The Fah(6287SB) allele is a missense mutation in exon 6, and Fah(5961SB) is a splice mutation causing loss of exon 7, a subsequent frameshift in the resulting mRNA, and a severe reduction of Fah mRNA levels. Increased levels of the diagnostic metabolite succinylacetone in the urine of the Fah(6287SB) and Fah(5961SB) mutants indicate that these mutations cause a decrease in Fah enzymatic activity. Thus, the neonatal phenotype present in both mutants is due to a deficiency in Fah caused by a point mutation, and we propose Fah(5961SB) and Fah(6287SB) as mouse models for acute and chronic forms of human HT1, respectively.
Figures
References
-
- Goldsmith L A, Laberge C. The Metabolic Basis of Inherited Disease. 6th Ed. Vol. 1. New York: McGraw–Hill; 1989. pp. 547–562.
-
- Berger R, van Faassen H, Smith G P A. Clin Chim Acta. 1983;134:129–141. - PubMed
-
- Overturf K, Al-Dhalimy M, Tanguay R, Brantly M, Ou C, Finegold M, Grompe M. Nat Genet. 1996;12:266–273. - PubMed
-
- Labelle Y, Phaneuf D, Leclerc B, Tanguay R M. Hum Mol Genet. 1993;2:941–946. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
