

Persistently modified h-channels after complex febrile seizures convert the seizure-induced enhancement of inhibition to hyperexcitability
- PMID: 11231632
- PMCID: PMC3382967
- DOI: 10.1038/85480
Persistently modified h-channels after complex febrile seizures convert the seizure-induced enhancement of inhibition to hyperexcitability
Abstract
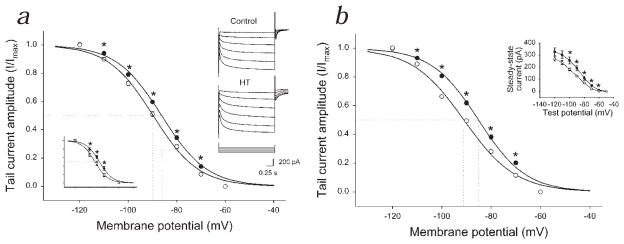
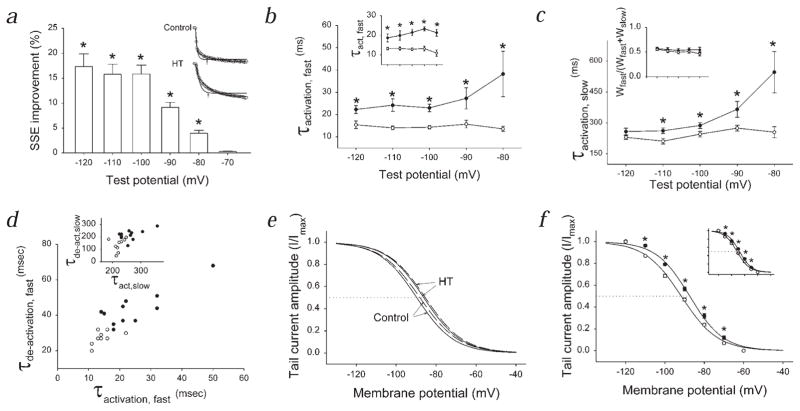
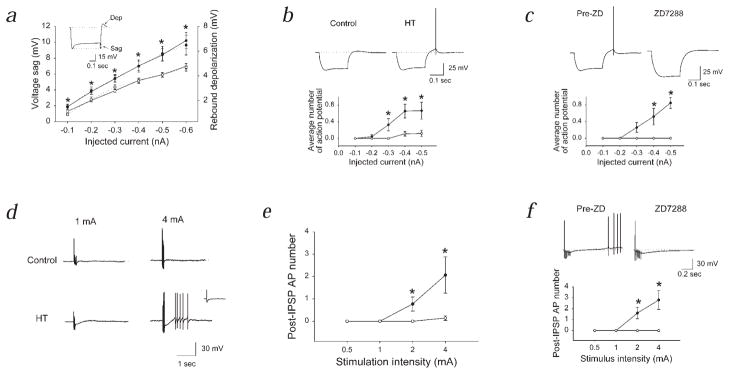
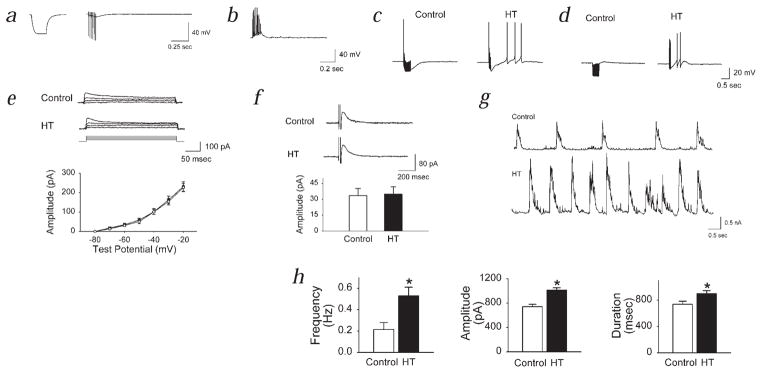
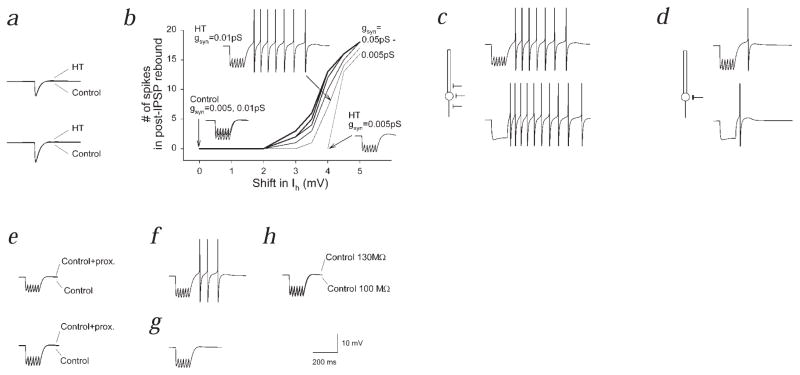
Febrile seizures are the most common type of developmental seizures, affecting up to 5% of children. Experimental complex febrile seizures involving the immature rat hippocampus led to a persistent lowering of seizure threshold despite an upregulation of inhibition. Here we provide a mechanistic resolution to this paradox by showing that, in the hippocampus of rats that had febrile seizures, the long-lasting enhancement of the widely expressed intrinsic membrane conductance Ih converts the potentiated synaptic inhibition to hyperexcitability in a frequency-dependent manner. The altered gain of this molecular inhibition-excitation converter reveals a new mechanism for controlling the balance of excitation-inhibition in the limbic system. In addition, here we show for the first time that h-channels are modified in a human neurological disease paradigm.
Figures
References
-
- Nusser Z, Hájos N, Somogyi P, Mody I. Increased number of synaptic GABA(A) receptors underlies potentiation at hippocampal inhibitory synapses. Nature. 1998;395:172–177. - PubMed
-
- Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA. Selective changes in single cell GABAA receptor subunit expression and function in temporal lobe epilepsy. Nature Med. 1998;4:1166–1172. - PubMed
-
- Buhl EH, Otis TS, Mody I. Zinc-induced collapse of augmented inhibition by GABA in a temporal lobe epilepsy model. Science. 1996;271:369–373. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
