

Activation of the murine EP3 receptor for PGE2 inhibits cAMP production and promotes platelet aggregation
- PMID: 11238561
- PMCID: PMC199422
- DOI: 10.1172/JCI10881
Activation of the murine EP3 receptor for PGE2 inhibits cAMP production and promotes platelet aggregation
Abstract
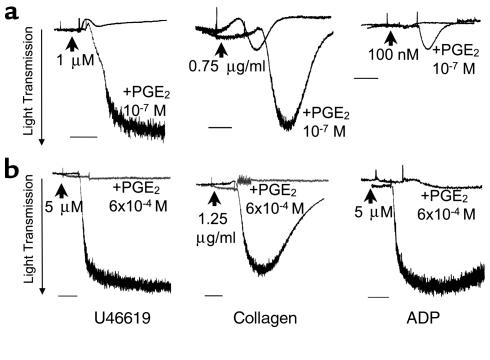
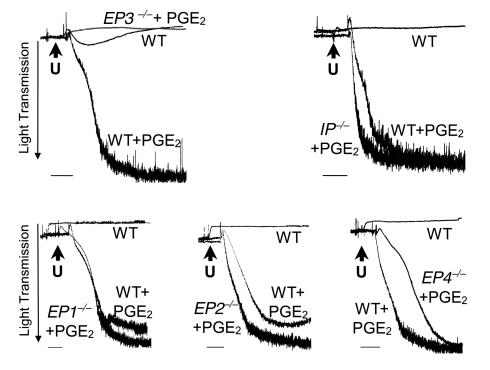
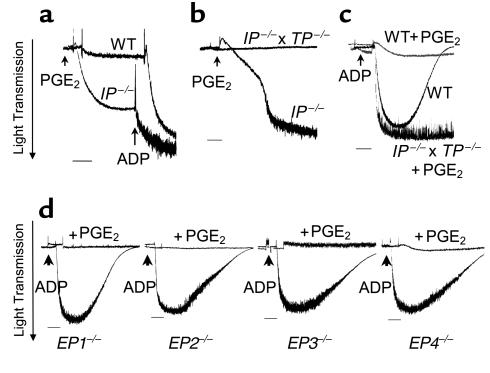
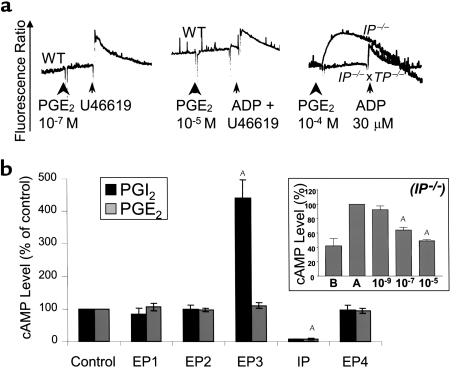
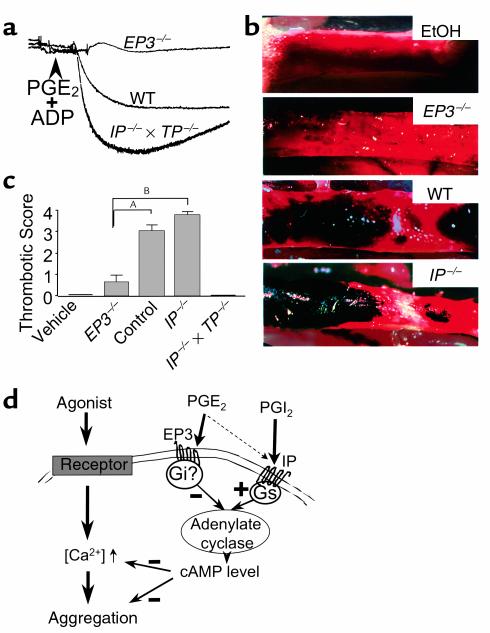
The importance of arachidonic acid metabolites (termed eicosanoids), particularly those derived from the COX-1 and COX-2 pathways (termed prostanoids), in platelet homeostasis has long been recognized. Thromboxane is a potent agonist, whereas prostacyclin is an inhibitor of platelet aggregation. In contrast, the effect of prostaglandin E2 (PGE2) on platelet aggregation varies significantly depending on its concentration. Low concentrations of PGE2 enhance platelet aggregation, whereas high PGE2 levels inhibit aggregation. The mechanism for this dual action of PGE2 is not clear. This study shows that among the four PGE2 receptors (EP1-EP4), activation of EP3 is sufficient to mediate the proaggregatory actions of low PGE2 concentration. In contrast, the prostacyclin receptor (IP) mediates the inhibitory effect of higher PGE2 concentrations. Furthermore, the relative activation of these two receptors, EP3 and IP, regulates the intracellular level of cAMP and in this way conditions the response of the platelet to aggregating agents. Consistent with these findings, loss of the EP3 receptor in a model of venous inflammation protects against formation of intravascular clots. Our results suggest that local production of PGE2 during an inflammatory process can modulate ensuing platelet responses.
Figures
References
-
- Smith WL. Prostaglandin biosynthesis and its compartmentation in vascular smooth muscle and endothelial cells. Annu Rev Physiol. 1986;48:251–262. - PubMed
-
- Bishop-Bailey D, et al. Induction of cyclooxygenase-2 in human saphenous vein and internal mammary artery. Arterioscler Thromb Vasc Biol. 1997;17:1644–1648. - PubMed
-
- Bishop-Bailey D, Pepper JR, Larkin SW, Mitchell JA. Differential induction of cyclooxygenase-2 in human arterial and venous smooth muscle: role of endogenous prostanoids. Arterioscler Thromb Vasc Biol. 1998;18:1655–1661. - PubMed
-
- Mitchell JA, Larkin S, Williams TJ. Cyclooxygenase-2: regulation and relevance in inflammation. Biochem Pharmacol. 1995;50:1535–1542. - PubMed
-
- Stemme V, Swedenborg J, Claesson H, Hansson GK. Expression of cyclo-oxygenase-2 in human atherosclerotic carotid arteries. Eur J Vasc Endovasc Surg. 2000;20:146–152. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
