

Essential role of nuclear factor (NF)-kappaB-inducing kinase and inhibitor of kappaB (IkappaB) kinase alpha in NF-kappaB activation through lymphotoxin beta receptor, but not through tumor necrosis factor receptor I
- PMID: 11238593
- PMCID: PMC2193391
- DOI: 10.1084/jem.193.5.631
Essential role of nuclear factor (NF)-kappaB-inducing kinase and inhibitor of kappaB (IkappaB) kinase alpha in NF-kappaB activation through lymphotoxin beta receptor, but not through tumor necrosis factor receptor I
Abstract
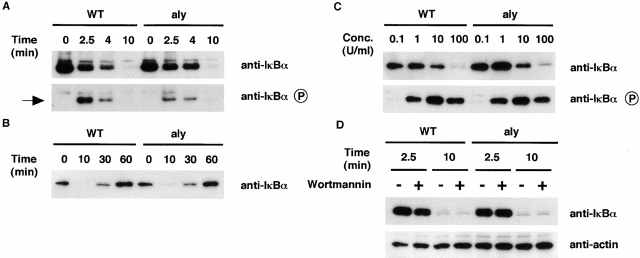
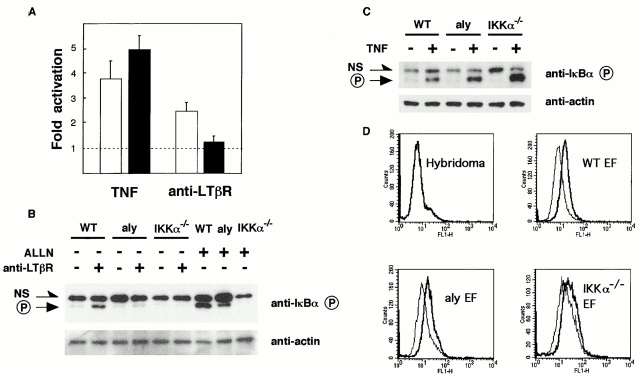
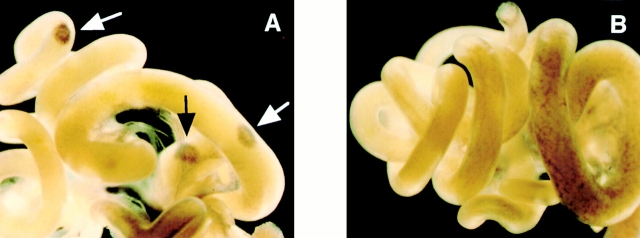
Both nuclear factor (NF)-kappaB-inducing kinase (NIK) and inhibitor of kappaB (IkappaB) kinase (IKK) have been implicated as essential components for NF-kappaB activation in response to many external stimuli. However, the exact roles of NIK and IKKalpha in cytokine signaling still remain controversial. With the use of in vivo mouse models, rather than with enforced gene-expression systems, we have investigated the role of NIK and IKKalpha in signaling through the type I tumor necrosis factor (TNF) receptor (TNFR-I) and the lymphotoxin beta receptor (LTbetaR), a receptor essential for lymphoid organogenesis. TNF stimulation induced similar levels of phosphorylation and degradation of IkappaBalpha in embryonic fibroblasts from either wild-type or NIK-mutant mice. In contrast, LTbetaR stimulation induced NF-kappaB activation in wild-type mice, but the response was impaired in embryonic fibroblasts from NIK-mutant and IKKalpha-deficient mice. Consistent with the essential role of IKKalpha in LTbetaR signaling, we found that development of Peyer's patches was defective in IKKalpha-deficient mice. These results demonstrate that both NIK and IKKalpha are essential for the induction of NF-kappaB through LTbetaR, whereas the NIK-IKKalpha pathway is dispensable in TNFR-I signaling.
Figures

References
-
- Ghosh S., May M.J., Kopp E.B. NF-κB and Rel proteinsevolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998;16:225–260. - PubMed
-
- Van Antwerp D.J., Martin S.J., Verma I.M., Green D.R. Inhibition of TNF-induced apoptosis by NF-κB. Trends Cell Biol. 1998;8:107–111. - PubMed
-
- Baldwin A.S. The NF-κB and IκB proteinsnew discoveries and insights. Annu. Rev. Immunol. 1996;14:649–681. - PubMed
-
- Maniatis T. Catalysis by a multiprotein IκB kinase complex. Science. 1997;278:818–819. - PubMed
-
- Stancovski I., Baltimore D. NF-κB activationthe IκB kinase revealed? Cell. 1997;91:299–302. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
