

Aberrant CFTR-dependent HCO3- transport in mutations associated with cystic fibrosis
- PMID: 11242048
- PMCID: PMC3943212
- DOI: 10.1038/35065099
Aberrant CFTR-dependent HCO3- transport in mutations associated with cystic fibrosis
Abstract
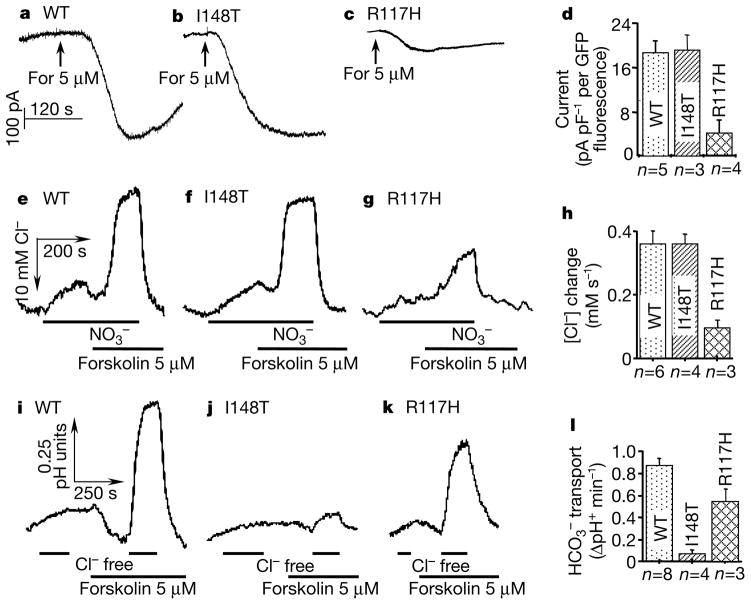
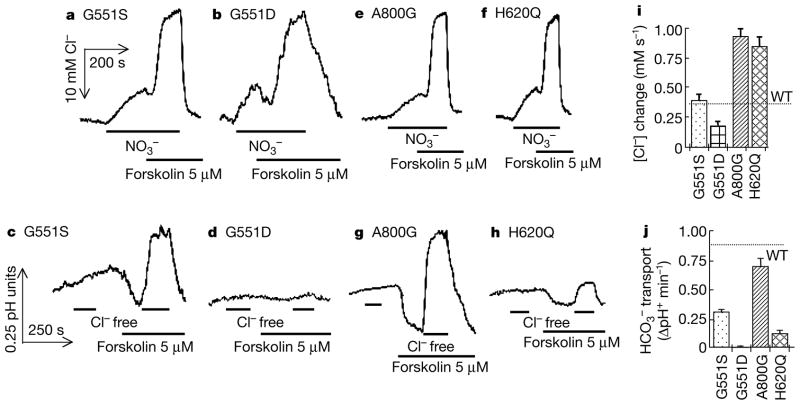
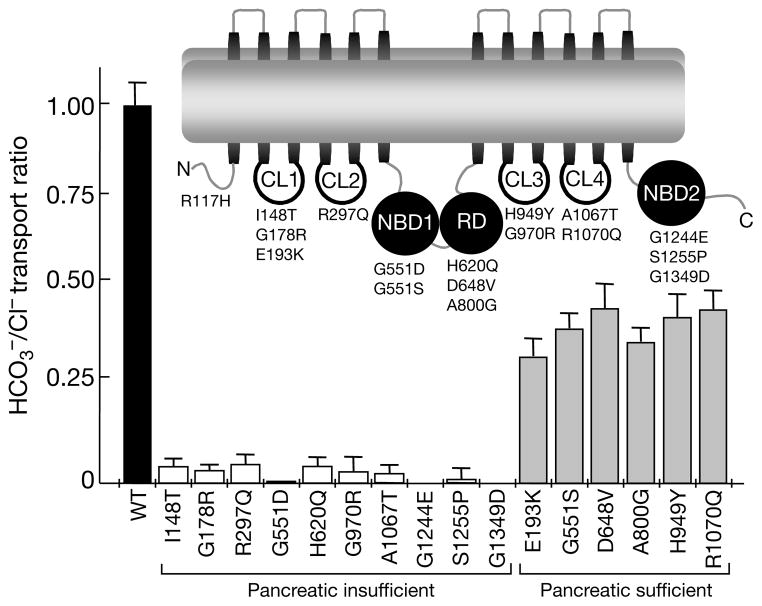
Cystic fibrosis (CF) is a disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR). Initially, Cl- conductance in the sweat duct was discovered to be impaired in CF, a finding that has been extended to all CFTR-expressing cells. Subsequent cloning of the gene showed that CFTR functions as a cyclic-AMP-regulated Cl- channel; and some CF-causing mutations inhibit CFTR Cl- channel activity. The identification of additional CF-causing mutants with normal Cl- channel activity indicates, however, that other CFTR-dependent processes contribute to the disease. Indeed, CFTR regulates other transporters, including Cl(-)-coupled HCO3- transport. Alkaline fluids are secreted by normal tissues, whereas acidic fluids are secreted by mutant CFTR-expressing tissues, indicating the importance of this activity. HCO3- and pH affect mucin viscosity and bacterial binding. We have examined Cl(-)-coupled HCO3- transport by CFTR mutants that retain substantial or normal Cl- channel activity. Here we show that mutants reported to be associated with CF with pancreatic insufficiency do not support HCO3- transport, and those associated with pancreatic sufficiency show reduced HCO3- transport. Our findings demonstrate the importance of HCO3- transport in the function of secretory epithelia and in CF.
Figures
References
-
- Quinton PM. Chloride impermeability in cystic fibrosis. Nature. 1983;301:421–422. - PubMed
-
- Quinton PM. Physiological basis of cystic fibrosis: a historical perspective. Physiol Rev. 1999;79:S3–S22. - PubMed
-
- Pilewski JM, Frizzell RA. Role of CFTR in airway disease. Physiol Rev. 1999;79:S215–S255. - PubMed
-
- Grubb BR, Boucher RC. Pathophysiology of gene-targeted mouse models for cystic fibrosis. Physiol Rev. 1999;79:S193–S213. - PubMed
-
- Rommens JM, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
