

Delayed neurodegeneration in neonatal rat thalamus after hypoxia-ischemia is apoptosis
- PMID: 11245678
- PMCID: PMC6762598
- DOI: 10.1523/JNEUROSCI.21-06-01931.2001
Delayed neurodegeneration in neonatal rat thalamus after hypoxia-ischemia is apoptosis
Abstract
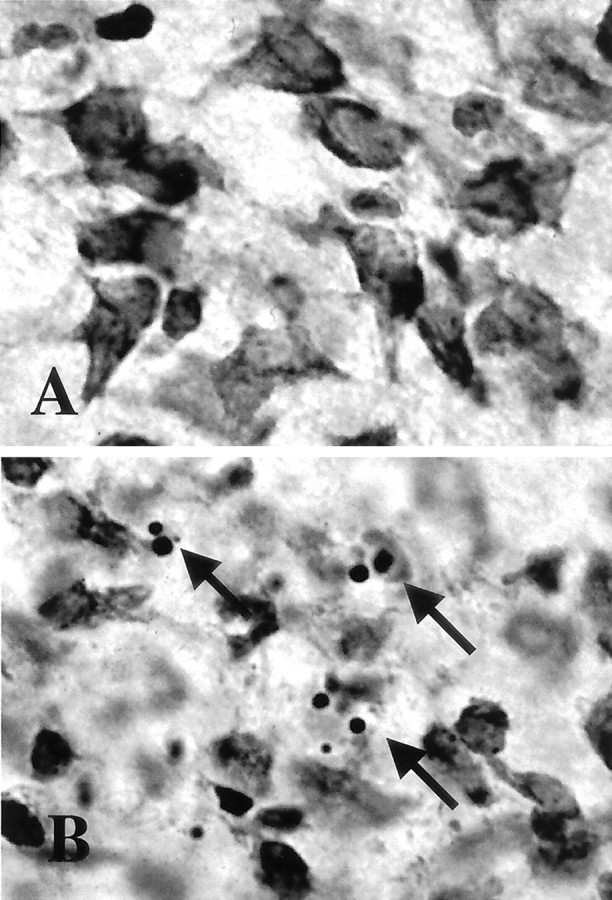
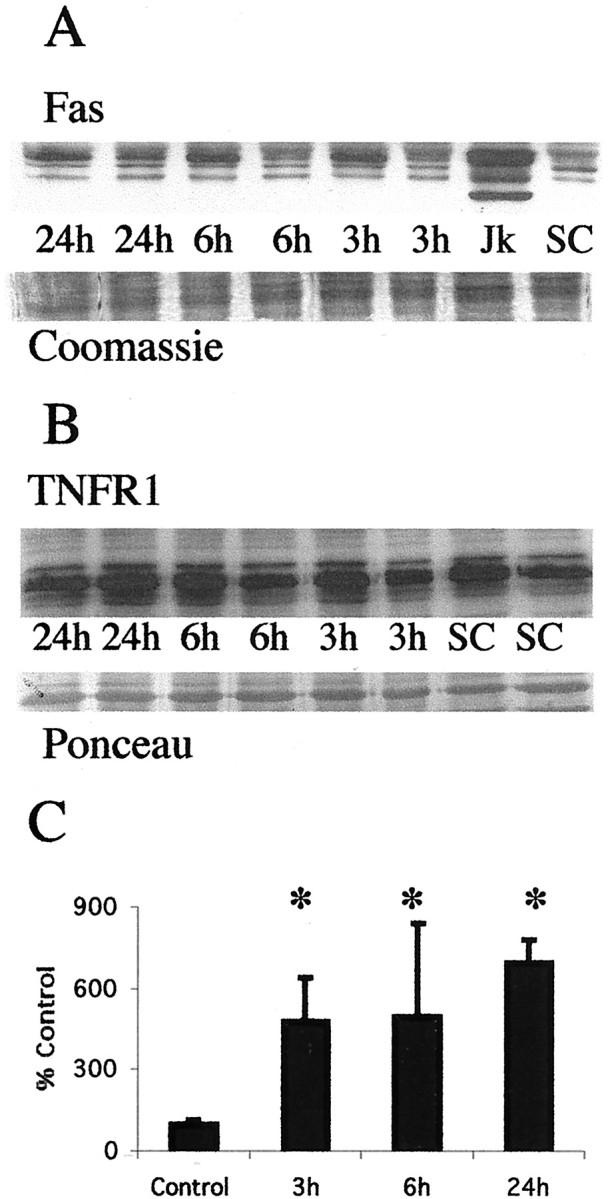
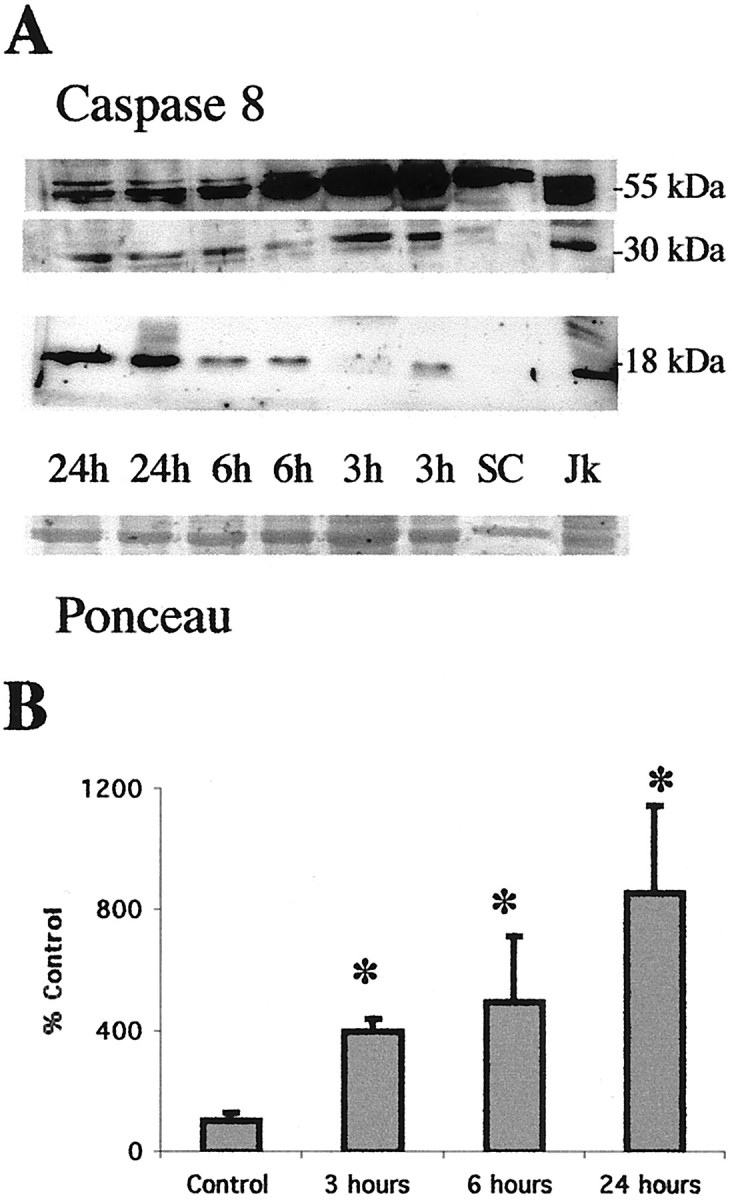
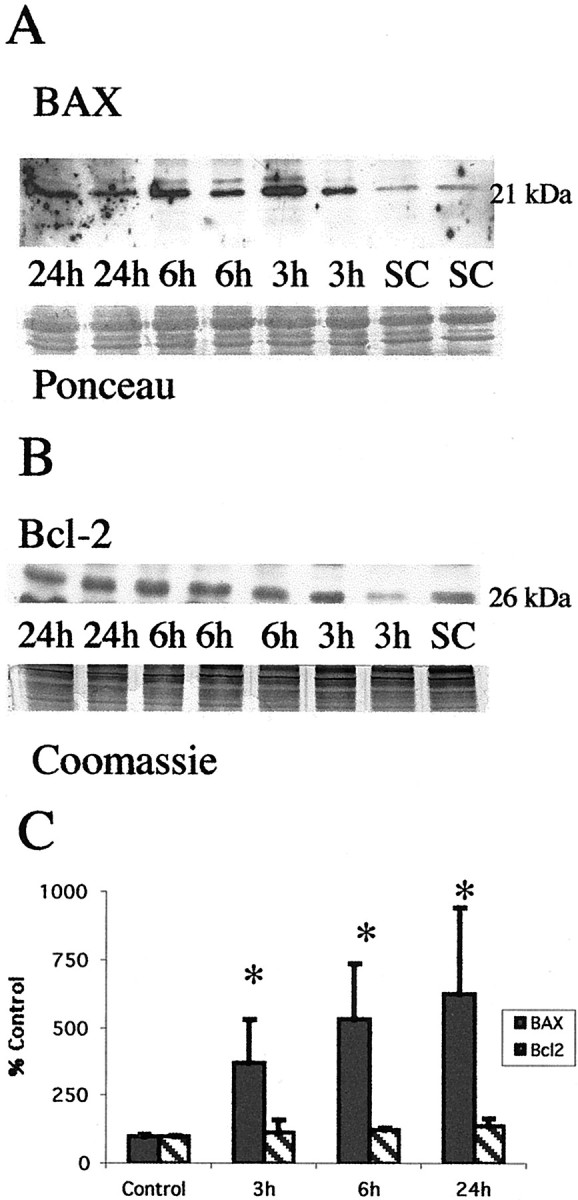
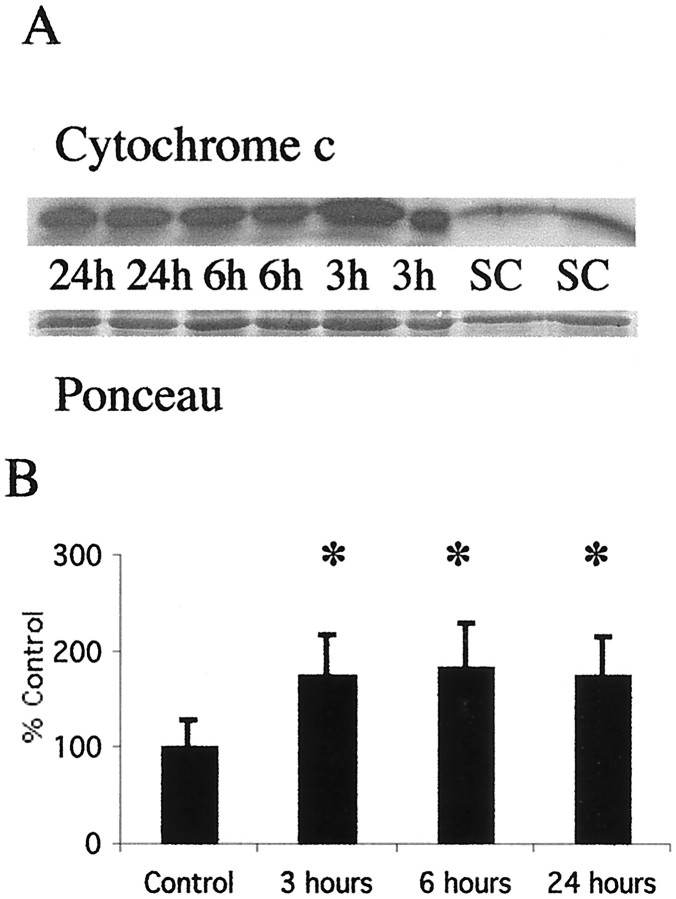
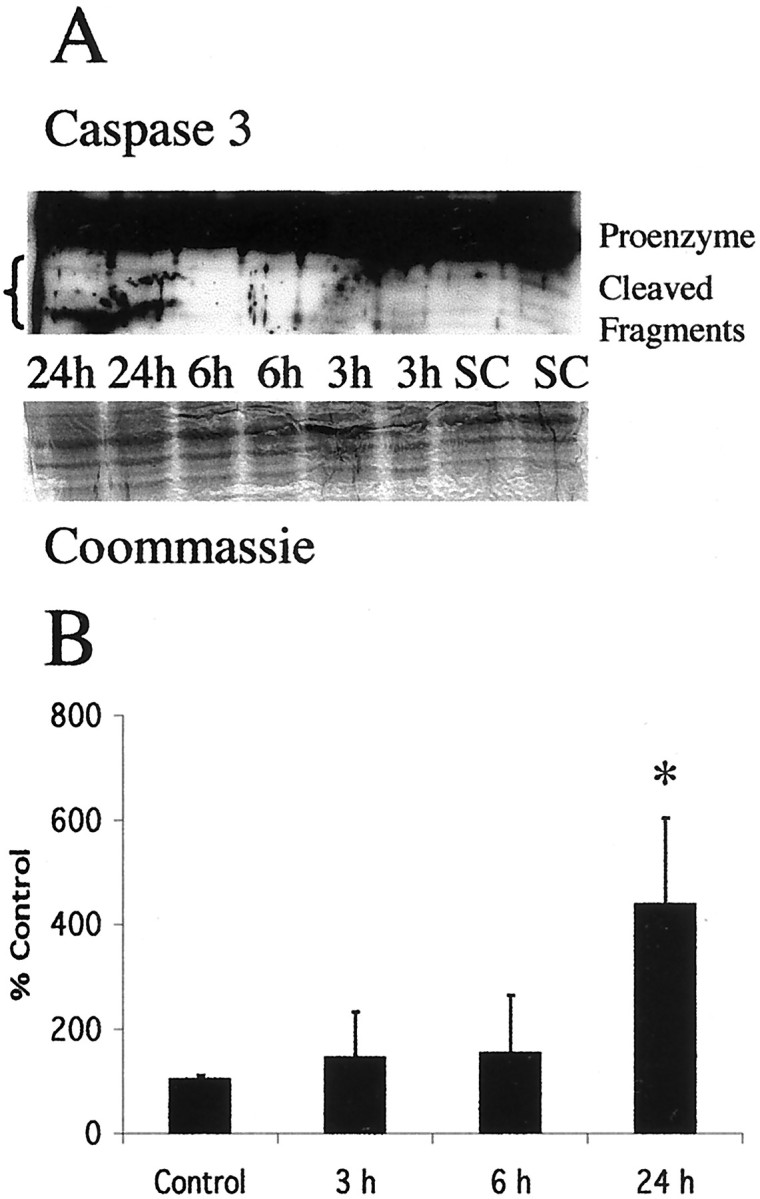
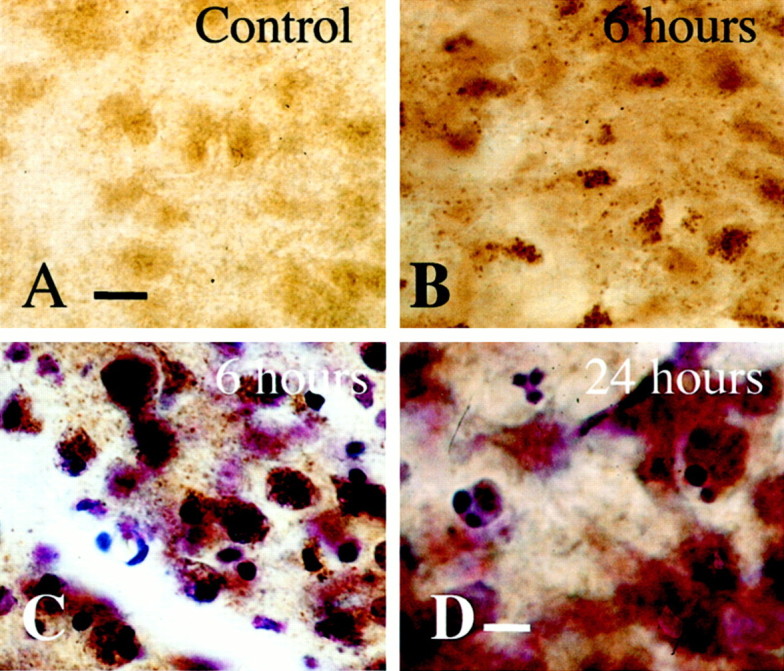
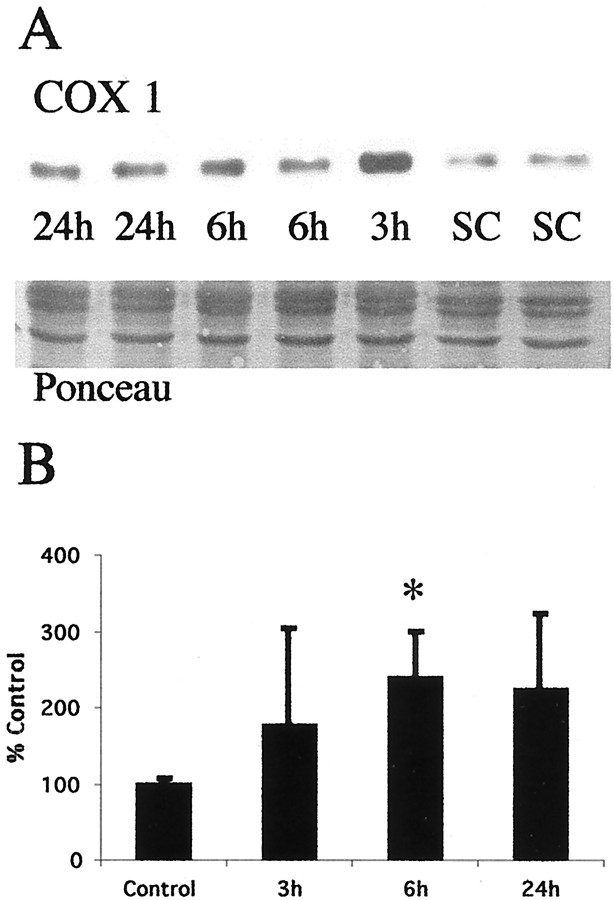
Brain injury in newborns can cause deficits in motor and sensory function. In most models of neonatal brain injury, thalamic damage often occurs. Using the Rice-Vannucci model of neonatal hypoxic-ischemic brain injury, we have shown that neuronal degeneration in somatosensory thalamus is delayed in onset ( approximately 24 hr) compared with cortical and striatal injury and exhibits prominent structural features of apoptosis. In the present study, we examined whether cell death in the thalamus has molecular features of apoptosis. Fas death receptor protein expression increased rapidly after neonatal hypoxia-ischemia, in concert with cleavage of procaspase 8 to its active form. Concurrently, the levels of Bax in mitochondrial-enriched cell fractions increase, and cytochrome c accumulates in the soluble fraction. Mitochondria accumulate in a perinuclear distribution by 6 hr after hypoxia-ischemia. Cytochrome oxidase subunit 1 protein levels also increase at 6 hr after hypoxia-ischemia. Increased levels of Fas death receptor, Bax, and cytochrome c, activation of caspase 8, and abnormalities in mitochondria in the thalamus significantly precede the activation of caspase 3 and the appearance of neuronal apoptosis at 24 hr. We conclude that the delayed neurodegeneration in neonatal rat ventral basal thalamus after hypoxic-ischemic injury is apoptosis mediated by death receptor activation.
Figures
References
-
- Al-Abdulla NA, Portera-Cailliau C, Martin LJ. Occipital cortex ablation in adult rat causes retrograde neuronal death in the lateral geniculate nucleus that resembles apoptosis. Neuroscience. 1998;86:191–209. - PubMed
-
- Almeida OF, Conde GL, Crochemore C, Demeneix BA, Fischer D, Hassan AH, Meyer M, Holsboer F, Michaelidis TM. Subtle shifts in the ratio between pro- and antiapoptotic molecules after activation of corticosteroid receptors decide neuronal fate. FASEB J. 2000;14:779–790. - PubMed
-
- Antonawich FJ, Federoff HJ, Davis JN. BCL-2 transduction, using a herpes simplex virus amplicon, protects hippocampal neurons from transient global ischemia. Exp Neurol. 1999;156:130–137. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous