

The influence of glutamate receptor 2 expression on excitotoxicity in Glur2 null mutant mice
- PMID: 11264298
- PMCID: PMC6762387
- DOI: 10.1523/JNEUROSCI.21-07-02224.2001
The influence of glutamate receptor 2 expression on excitotoxicity in Glur2 null mutant mice
Abstract
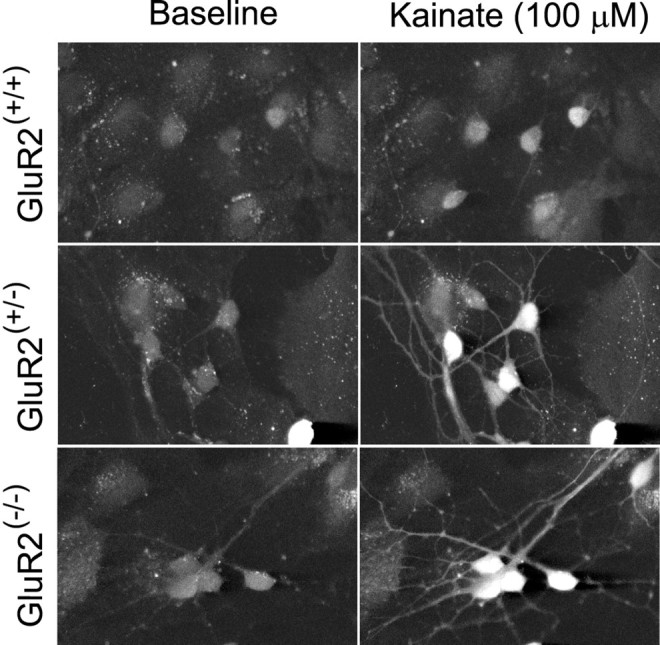
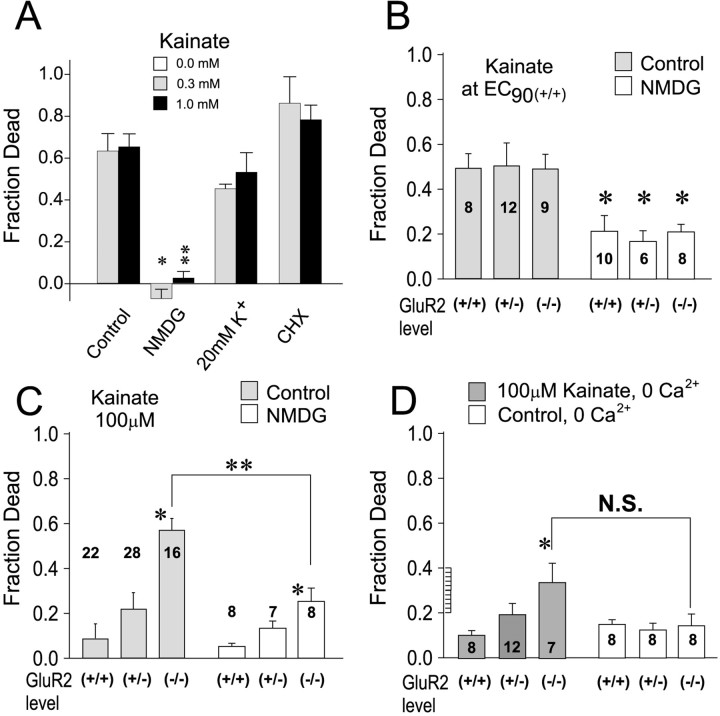
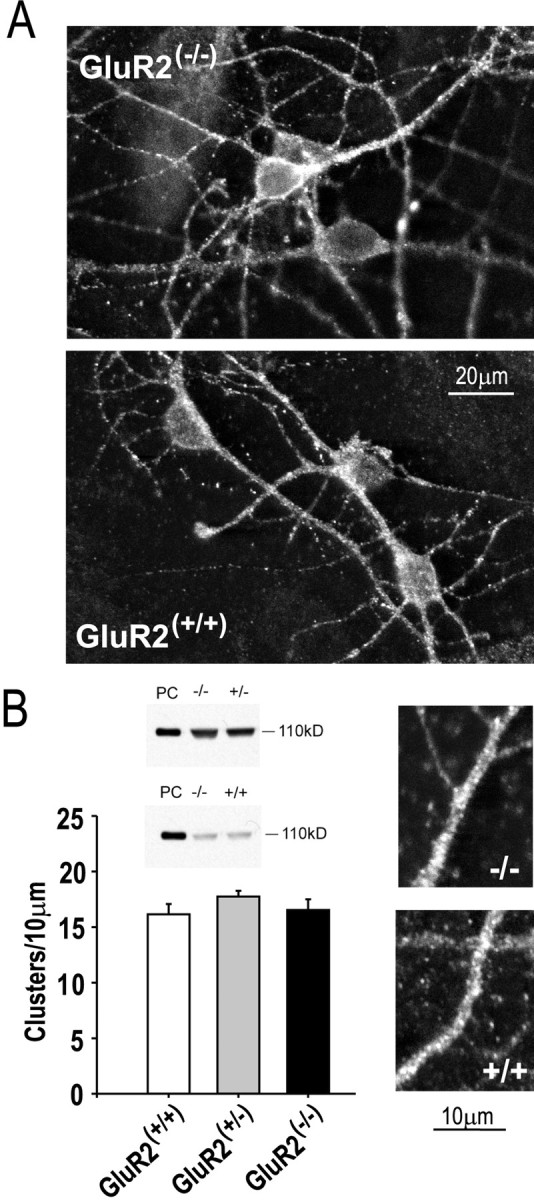
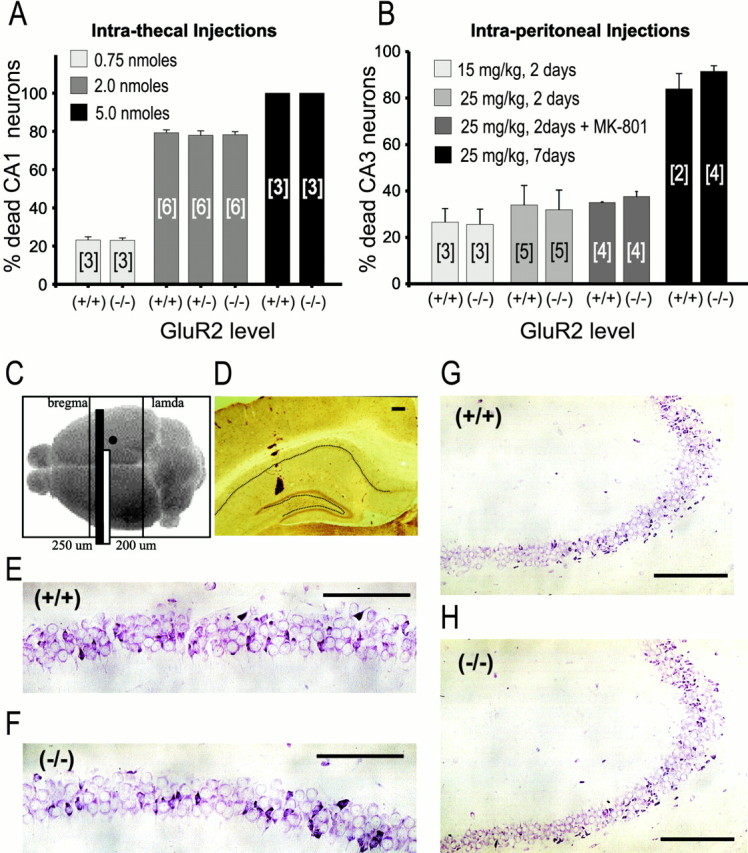
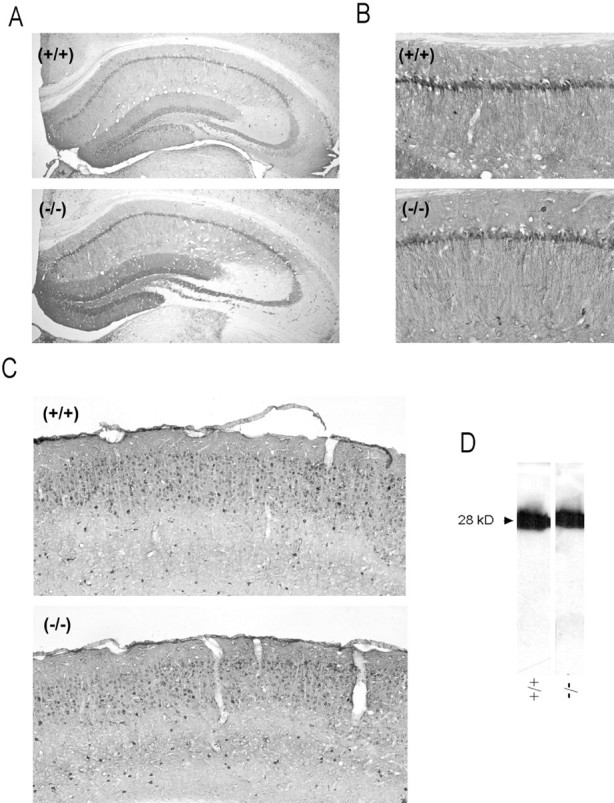
AMPA receptor (AMPAR)-mediated ionic currents that govern gene expression, synaptic strength, and plasticity also can trigger excitotoxicity. However, native AMPARs exhibit heterogeneous pharmacological, biochemical, and ionic permeability characteristics, which are governed partly by receptor subunit composition. Consequently, the mechanisms governing AMPAR-mediated excitotoxicity have been difficult to elucidate. The GluR2 subunit is of particular interest because it influences AMPAR pharmacology, Ca(2+) permeability, and AMPAR interactions with intracellular proteins. In this paper we used mutant mice lacking the AMPAR subunit GluR2 to study AMPAR-mediated excitotoxicity in cultured cortical neurons and in hippocampal neurons in vivo. We examined the hypothesis that in these mice the level of GluR2 expression governs the vulnerability of neurons to excitotoxicity and further examined the ionic mechanisms that are involved. In cortical neuronal cultures AMPAR-mediated neurotoxicity paralleled the magnitude of kainate-evoked AMPAR-mediated currents, which were increased in neurons lacking GluR2. Ca(2+) permeability, although elevated in GluR2-deficient neurons, did not correlate with excitotoxicity. However, toxicity was reduced by removal of extracellular Na(+), the main charge carrier of AMPAR-mediated currents. In vivo, the vulnerability of CA1 hippocampal neurons to stereotactic kainate injections and of CA3 neurons to intraperitoneal kainate administration was independent of GluR2 level. Neurons lacking the GluR2 subunit did not demonstrate compensatory changes in the distribution, expression, or function of AMPARs or of Ca(2+)-buffering proteins. Thus GluR2 level may influence excitotoxicity by effects additional to those on Ca(2+) permeability, such as effects on agonist potency, ionic currents, and synaptic reorganization.
Figures
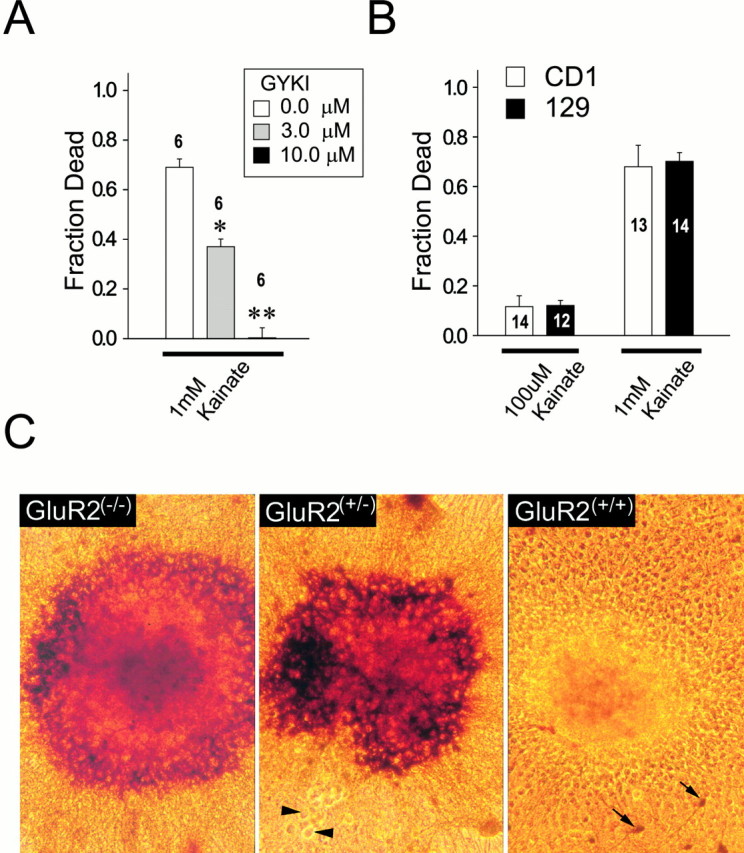
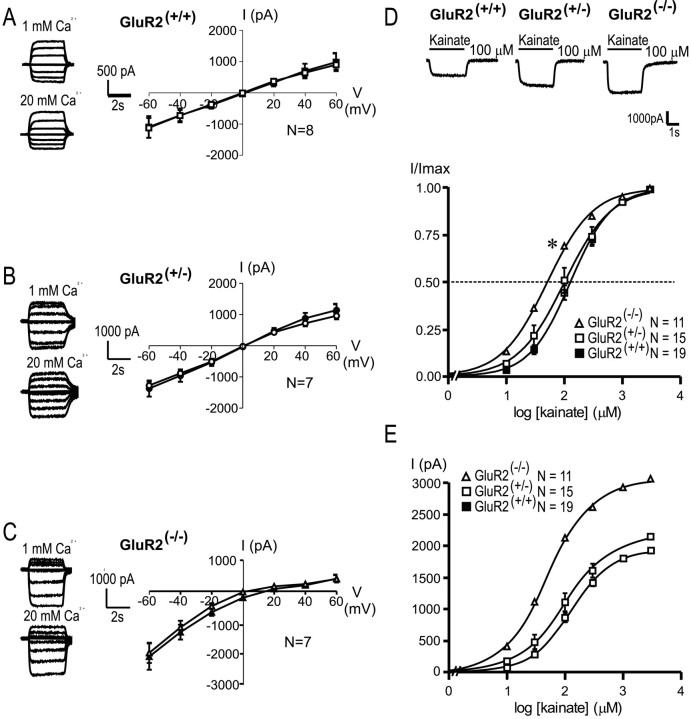
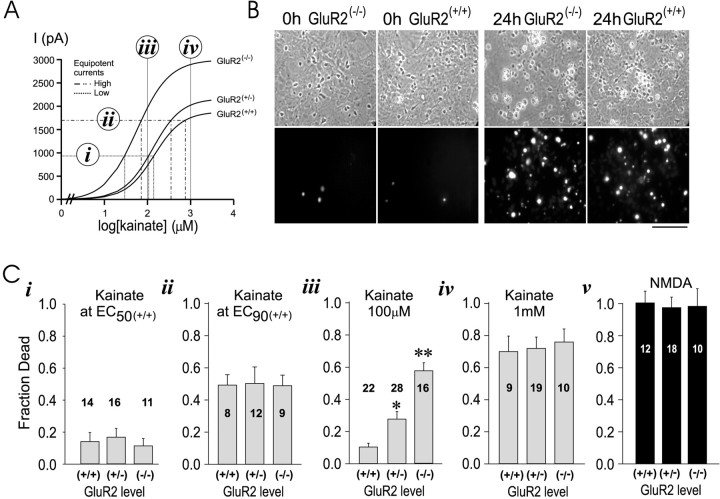
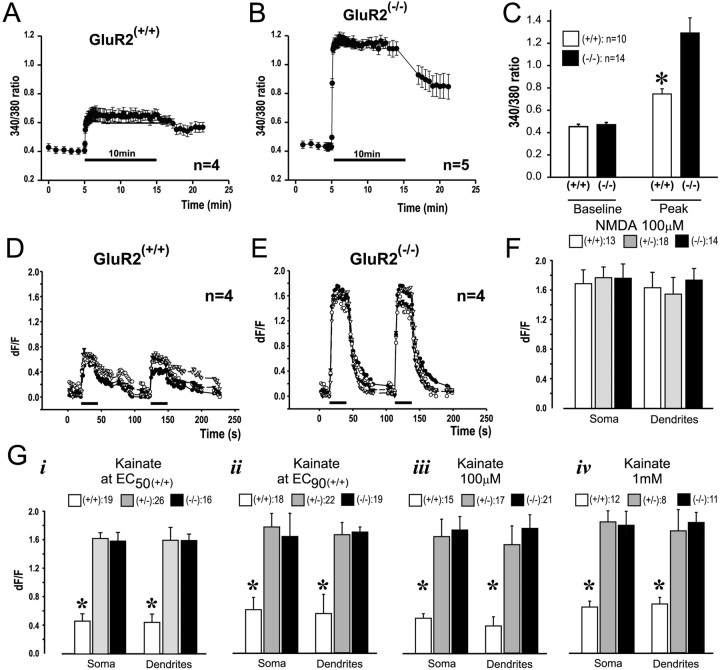
References
-
- Ben-Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14:375–403. - PubMed
-
- Bennett MV, Pellegrini-Giampietro DE, Gorter JA, Aronica E, Connor JA, Zukin RS. The GluR2 hypothesis: Ca2+-permeable AMPA receptors in delayed neurodegeneration. Cold Spring Harb Symp Quant Biol. 1996;61:373–384. - PubMed
-
- Blaustein MP. Calcium transport and buffering in neurons. Trends Neurosci. 1988;11:438–443. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous