

A comparative genomics approach to prediction of new members of regulons
- PMID: 11282972
- PMCID: PMC311042
- DOI: 10.1101/gr.149301
A comparative genomics approach to prediction of new members of regulons
Abstract
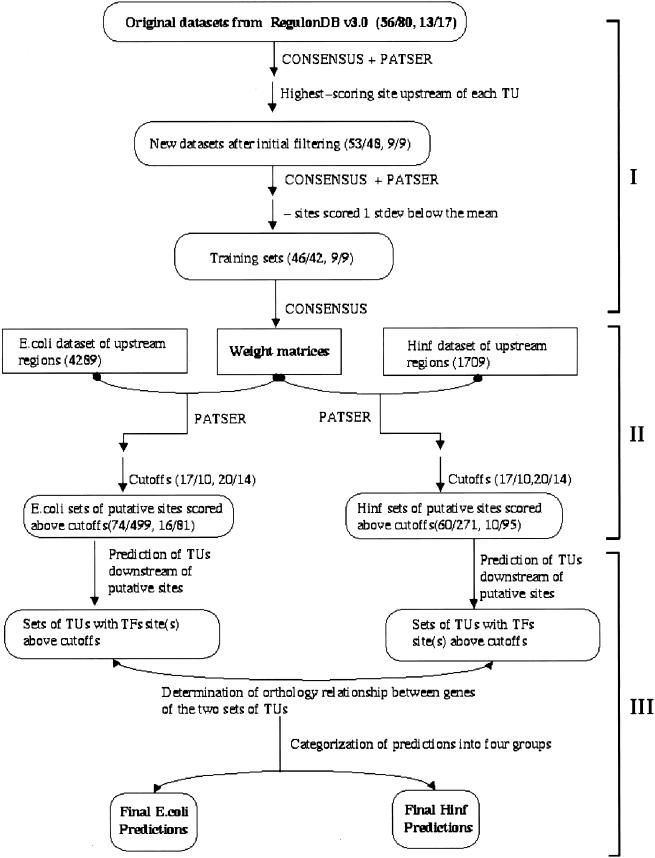
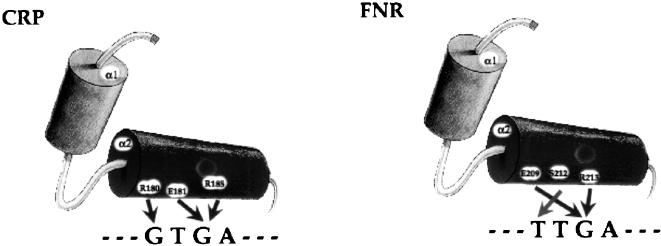
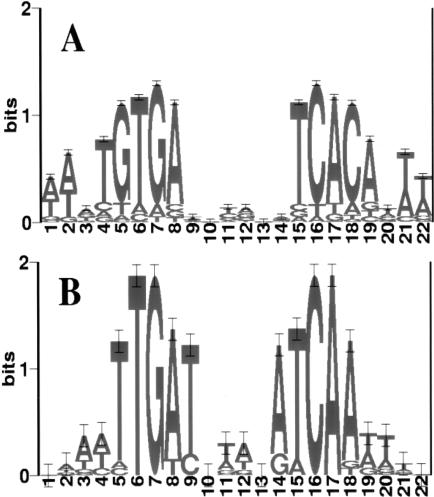
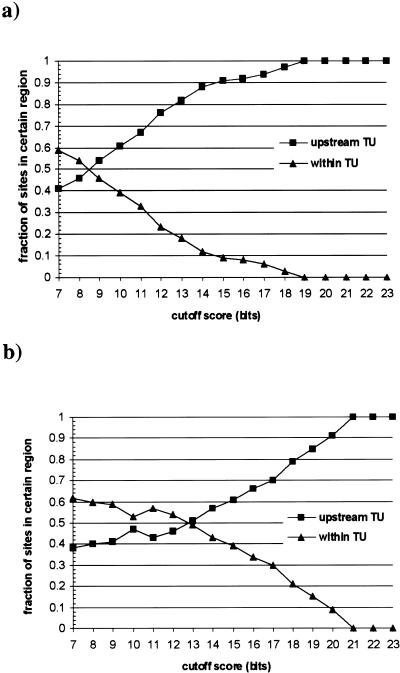
Identifying the complete transcriptional regulatory network for an organism is a major challenge. For each regulatory protein, we want to know all the genes it regulates, that is, its regulon. Examples of known binding sites can be used to estimate the binding specificity of the protein and to predict other binding sites. However, binding site predictions can be unreliable because determining the true specificity of the protein is difficult because of the considerable variability of binding sites. Because regulatory systems tend to be conserved through evolution, we can use comparisons between species to increase the reliability of binding site predictions. In this article, an approach is presented to evaluate the computational predictions of regulatory sites. We combine the prediction of transcription units having orthologous genes with the prediction of transcription factor binding sites based on probabilistic models. We augment the sets of genes in Escherichia coli that are expected to be regulated by two transcription factors, the cAMP receptor protein and the fumarate and nitrate reduction regulatory protein, through a comparison with the Haemophilus influenzae genome. At the same time, we learned more about the regulatory networks of H. influenzae, a species with much less experimental knowledge than E. coli. By studying orthologous genes subject to regulation by the same transcription factor, we also gained understanding of the evolution of the entire regulatory systems.
Figures





References
-
- Blattner FR, Plunkett G, III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. - PubMed
-
- Craven M, Page D, Shavlik J, Bockhorst J, Glasner J. A probabilistic learning approach to whole-genome operon prediction. Proc Int Conf Intell Syst Mol Biol. 2000;8:116–127. .. - PubMed
-
- Dandekar T, Snel B, Huynen M, Bork P. Conservation of gene order: A fingerprint of proteins that physically interact. Trends Biochem Sci. 1998;23:324–328. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases