

Caspase cleavage enhances the apoptosis-inducing effects of BAD
- PMID: 11287608
- PMCID: PMC86931
- DOI: 10.1128/MCB.21.9.3025-3036.2001
Caspase cleavage enhances the apoptosis-inducing effects of BAD
Abstract
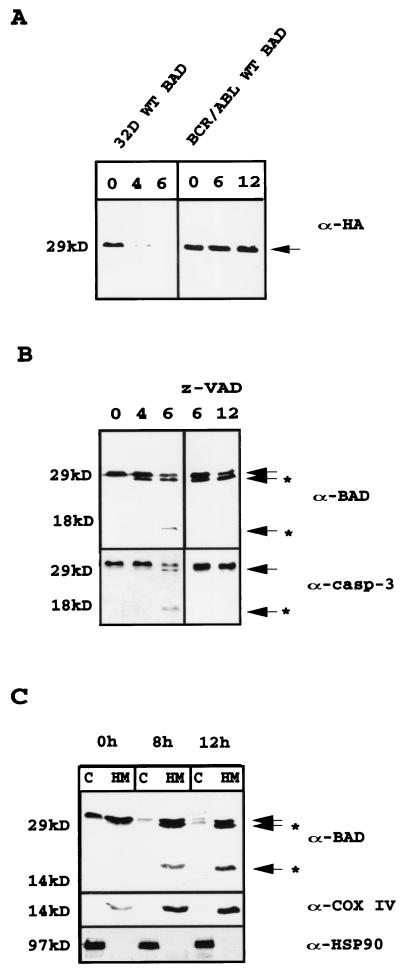
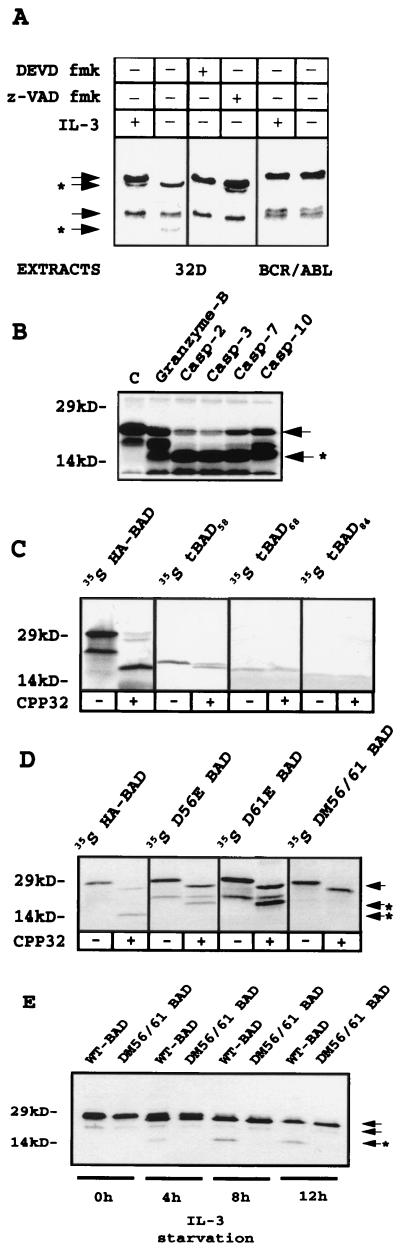
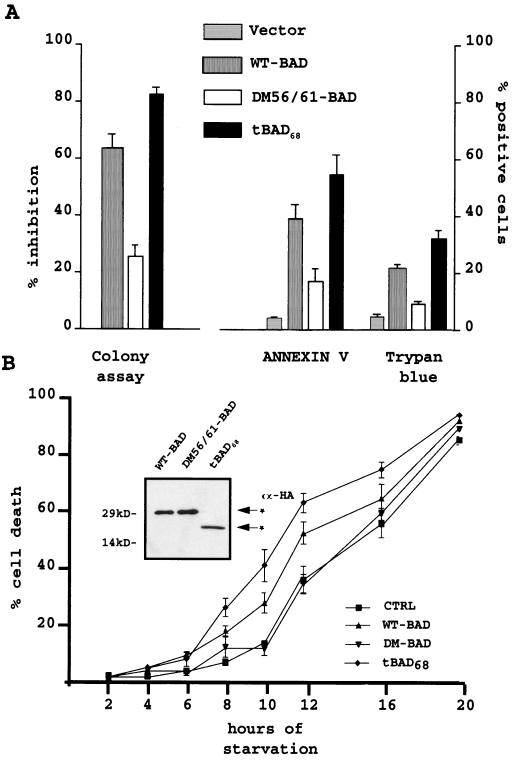
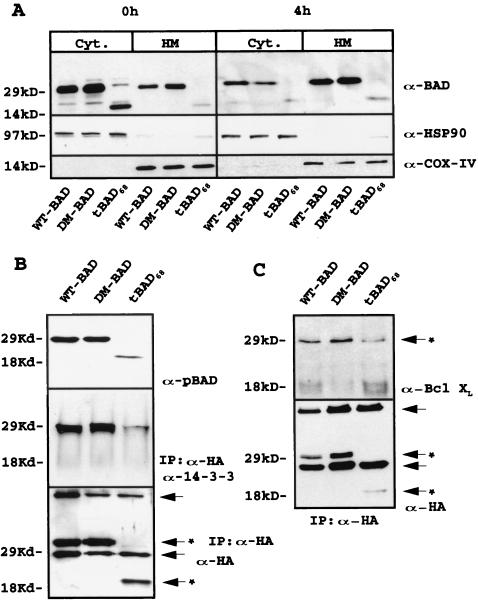
The function of BAD, a proapoptotic member of the Bcl-2 family, is regulated primarily by rapid changes in phosphorylation that modulate its protein-protein interactions and subcellular localization. We show here that, during interleukin-3 (IL-3) deprivation-induced apoptosis of 32Dcl3 murine myeloid precursor cells, BAD is cleaved by a caspase(s) at its N terminus to generate a 15-kDa truncated protein. The 15-kDa truncated BAD is a more potent inducer of apoptosis than the wild-type protein, whereas a mutant BAD resistant to caspase 3 cleavage is a weak apoptosis inducer. Truncated BAD is detectable only in the mitochondrial fraction, interacts with BCL-X(L) at least as effectively as the wild-type protein, and is more potent than wild-type BAD in inducing cytochrome c release. Human BAD, which is 43 amino acids shorter than its mouse counterpart, is also cleaved by a caspase(s) upon exposure of Jurkat T cells to anti-FAS antibody, tumor necrosis factor alpha (TNF-alpha), or TRAIL. Moreover, a truncated form of human BAD lacking the N-terminal 28 amino acids is more potent than wild-type BAD in inducing apoptosis. The generation of truncated BAD was blocked by Bcl-2 in IL-3-deprived 32Dcl3 cells but not in Jurkat T cells exposed to anti-FAS antibody, TNF-alpha, or TRAIL. Together, these findings point to a novel and important role for BAD in maintaining the apoptotic phenotype in response to various apoptosis inducers.
Figures

References
-
- Adams J M, Cory S. The Bc1–2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. - PubMed
-
- Chandler J M, Cohen G M, MacFarlane M. Different subcellular distribution of caspase-3 and caspase-7 following Fas-induced apoptosis in mouse liver. J Biol Chem. 1998;273:10815–10818. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous