

RNA polymerase III transcription complexes on chromosomal 5S rRNA genes in vivo: TFIIIB occupancy and promoter opening
- PMID: 11287621
- PMCID: PMC86947
- DOI: 10.1128/MCB.21.9.3166-3178.2001
RNA polymerase III transcription complexes on chromosomal 5S rRNA genes in vivo: TFIIIB occupancy and promoter opening
Abstract
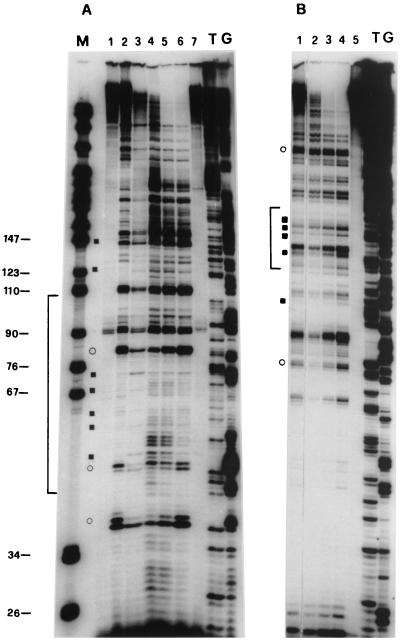
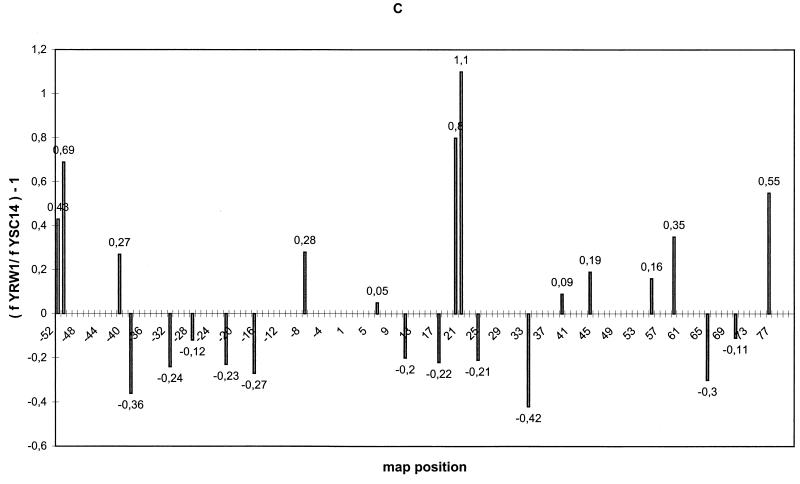
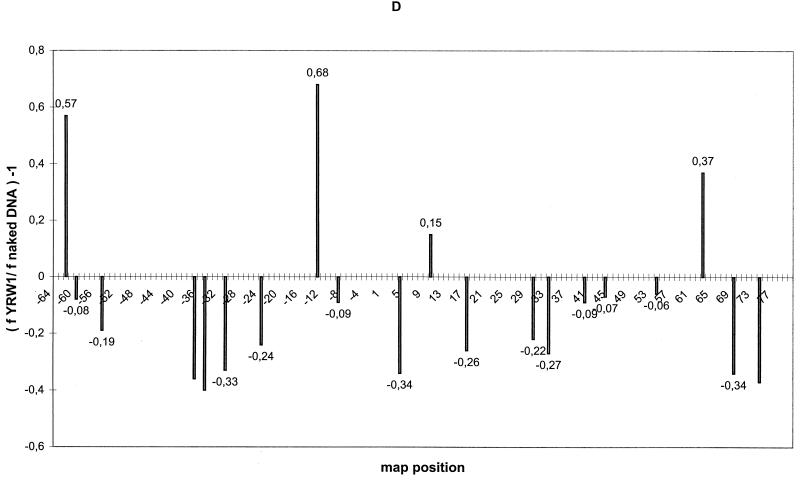
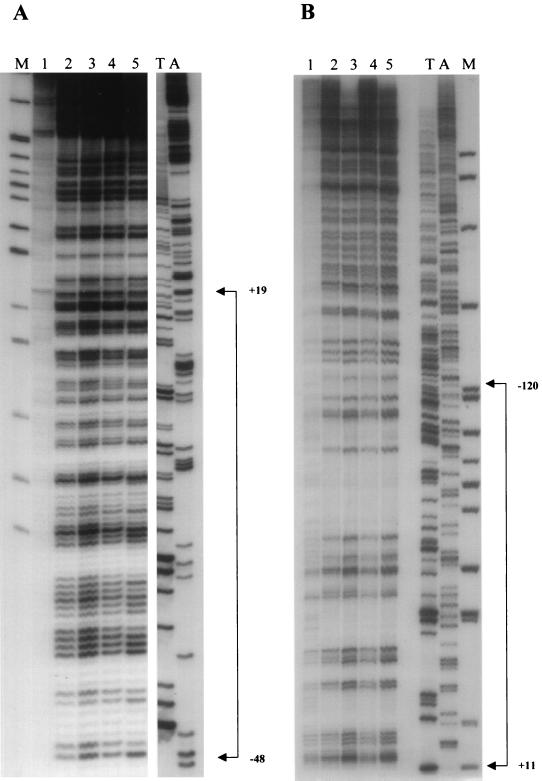
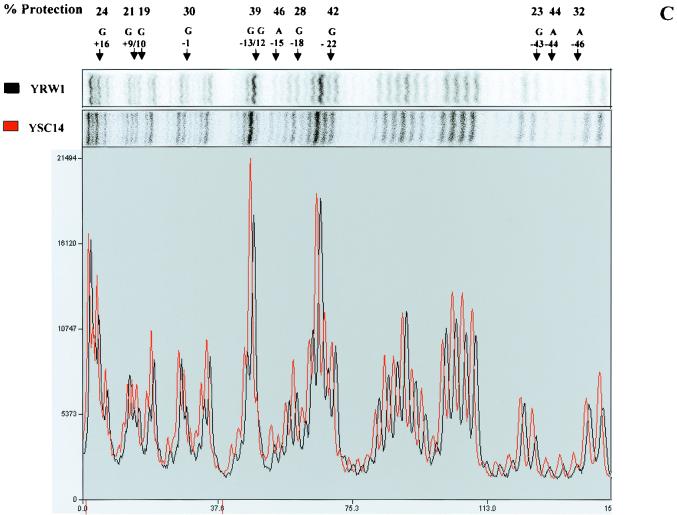
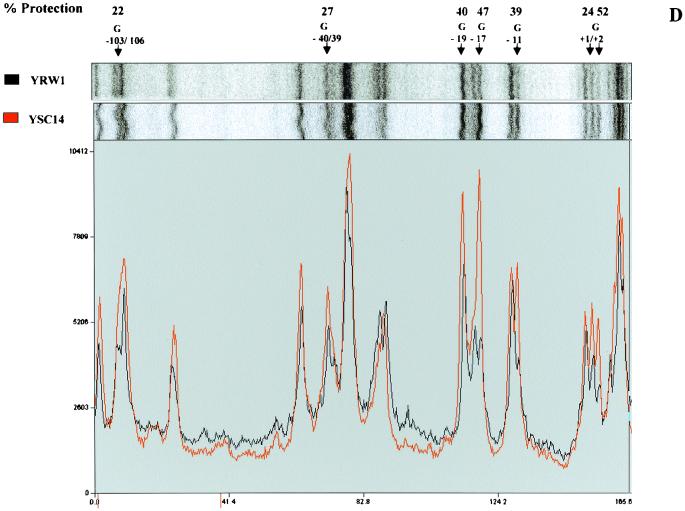
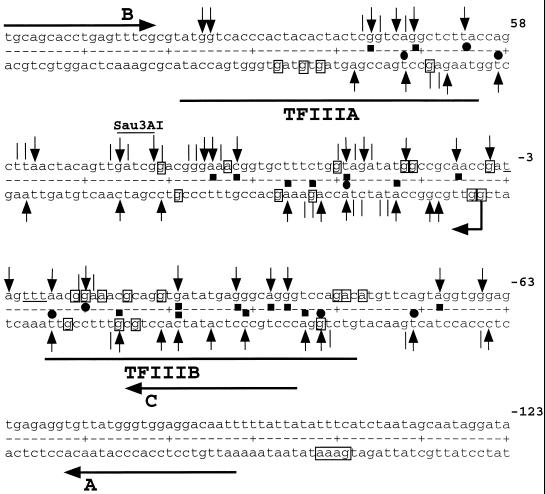
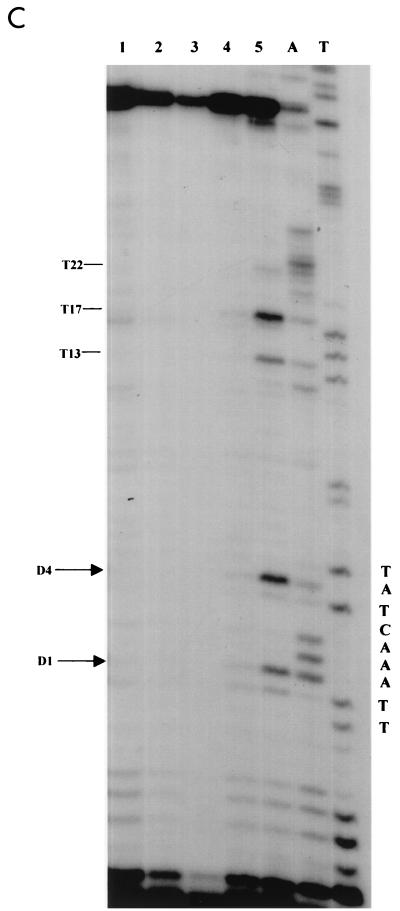
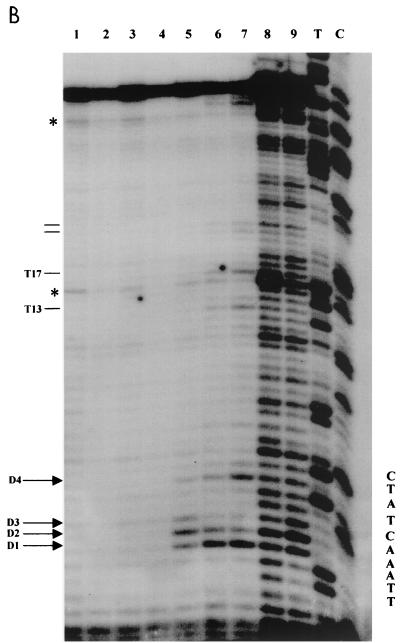
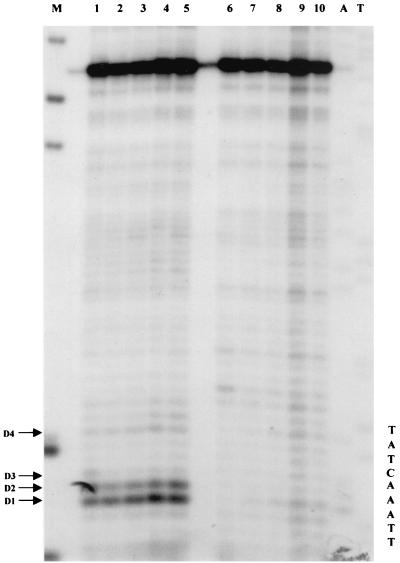
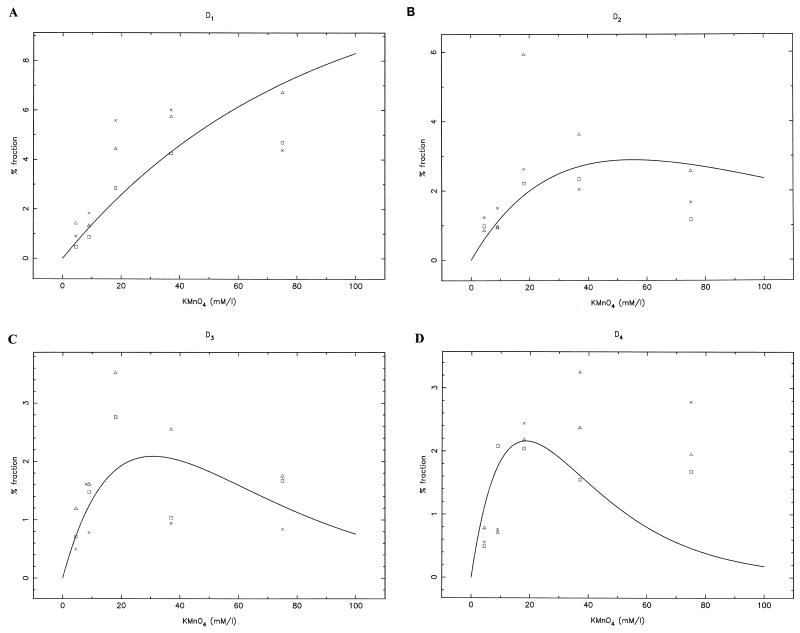
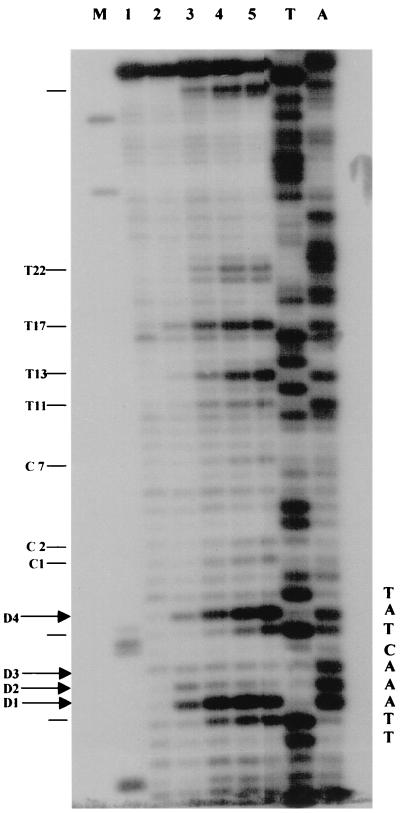
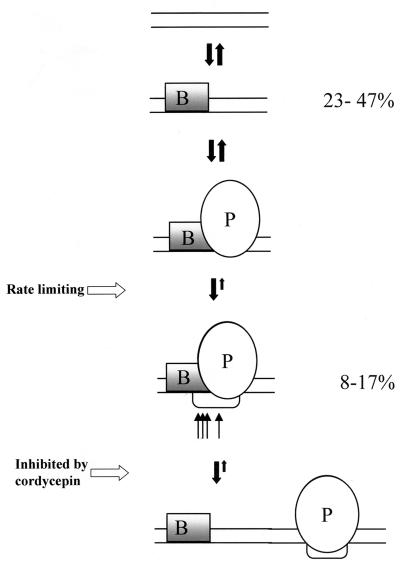
Quantitative analysis of multiple-hit potassium permanganate (KMnO(4)) footprinting has been carried out in vivo on Saccharomyces cerevisiae 5S rRNA genes. The results fix the number of open complexes at steady state in exponentially growing cells at between 8 and 17% of the 150 to 200 chromosomal copies. UV and dimethyl sulfate footprinting set the transcription factor TFIIIB occupancy at 23 to 47%. The comparison between the two values suggests that RNA polymerase III binding or promoter opening is the rate-limiting step in 5S rRNA transcription in vivo. Inhibition of RNA elongation in vivo by cordycepin confirms this result. An experimental system that is capable of providing information on the mechanistic steps involved in regulatory events in S. cerevisiae cells has been established.
Figures
Similar articles
-
S. cerevisiae TFIIIB is the transcription initiation factor proper of RNA polymerase III, while TFIIIA and TFIIIC are assembly factors.Cell. 1990 Jan 26;60(2):235-45. doi: 10.1016/0092-8674(90)90739-2. Cell. 1990. PMID: 2404611
-
Bending of the Saccharomyces cerevisiae 5S rRNA gene in transcription factor complexes.J Biol Chem. 1992 Nov 5;267(31):22562-9. J Biol Chem. 1992. PMID: 1429605
-
The RNA polymerase III transcription initiation factor TFIIIB participates in two steps of promoter opening.EMBO J. 2001 Jun 1;20(11):2823-34. doi: 10.1093/emboj/20.11.2823. EMBO J. 2001. PMID: 11387215 Free PMC article.
-
Yeast RNA polymerase III: transcription complexes and RNA synthesis.Braz J Med Biol Res. 1995 Feb;28(2):147-59. Braz J Med Biol Res. 1995. PMID: 7581036 Review.
-
Non-coding RNA production by RNA polymerase III is implicated in cancer.Nat Rev Cancer. 2008 Dec;8(12):911-4. doi: 10.1038/nrc2539. Epub 2008 Nov 6. Nat Rev Cancer. 2008. PMID: 18987635 Review.
Cited by
-
Applications of different weighting schemes to improve pathway-based analysis.Comp Funct Genomics. 2011;2011:463645. doi: 10.1155/2011/463645. Epub 2011 May 22. Comp Funct Genomics. 2011. PMID: 21687588 Free PMC article.
-
Visual analysis of the yeast 5S rRNA gene transcriptome: regulation and role of La protein.Mol Cell Biol. 2008 Jul;28(14):4576-87. doi: 10.1128/MCB.00127-08. Epub 2008 May 12. Mol Cell Biol. 2008. PMID: 18474615 Free PMC article.
-
MicroRNA: Biogenesis, Function and Role in Cancer.Curr Genomics. 2010 Nov;11(7):537-61. doi: 10.2174/138920210793175895. Curr Genomics. 2010. PMID: 21532838 Free PMC article.
-
Absolute gene occupancies by RNA polymerase III, TFIIIB, and TFIIIC in Saccharomyces cerevisiae.J Biol Chem. 2008 Sep 26;283(39):26568-76. doi: 10.1074/jbc.M803769200. Epub 2008 Jul 30. J Biol Chem. 2008. PMID: 18667429 Free PMC article.
-
Human U2 snRNA genes exhibit a persistently open transcriptional state and promoter disassembly at metaphase.Mol Cell Biol. 2008 Jun;28(11):3573-88. doi: 10.1128/MCB.00087-08. Epub 2008 Mar 31. Mol Cell Biol. 2008. PMID: 18378697 Free PMC article.
References
-
- Bartholomew B, Braun B R, Kassavetis G A, Geiduschek E P. Probing close DNA contacts of RNA polymerase III transcription complexes with the photoreactive nucleoside 4-thiodeoxythymidine. J Biol Chem. 1994;269:18090–18095. - PubMed
-
- Becker M M, Wang J C. Use of light for footprinting DNA in vivo. Nature. 1984;309:682–687. - PubMed
-
- Becker M M, Wang Z. Visualising intimate protein-DNA contacts and altered DNA structures with ultraviolet light. Biomethods. 1991;5:59–69.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases