

PartsList: a web-based system for dynamically ranking protein folds based on disparate attributes, including whole-genome expression and interaction information
- PMID: 11292848
- PMCID: PMC31319
- DOI: 10.1093/nar/29.8.1750
PartsList: a web-based system for dynamically ranking protein folds based on disparate attributes, including whole-genome expression and interaction information
Abstract
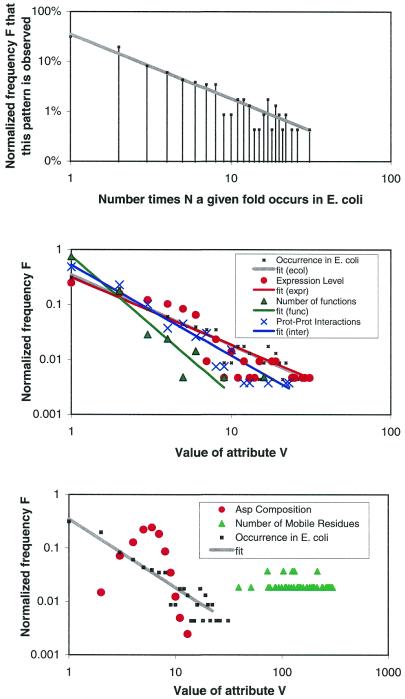
As the number of protein folds is quite limited, a mode of analysis that will be increasingly common in the future, especially with the advent of structural genomics, is to survey and re-survey the finite parts list of folds from an expanding number of perspectives. We have developed a new resource, called PartsList, that lets one dynamically perform these comparative fold surveys. It is available on the web at http://bioinfo.mbb.yale.edu/partslist and http://www.partslist.org. The system is based on the existing fold classifications and functions as a form of companion annotation for them, providing 'global views' of many already completed fold surveys. The central idea in the system is that of comparison through ranking; PartsList will rank the approximately 420 folds based on more than 180 attributes. These include: (i) occurrence in a number of completely sequenced genomes (e.g. it will show the most common folds in the worm versus yeast); (ii) occurrence in the structure databank (e.g. most common folds in the PDB); (iii) both absolute and relative gene expression information (e.g. most changing folds in expression over the cell cycle); (iv) protein-protein interactions, based on experimental data in yeast and comprehensive PDB surveys (e.g. most interacting fold); (v) sensitivity to inserted transposons; (vi) the number of functions associated with the fold (e.g. most multi-functional folds); (vii) amino acid composition (e.g. most Cys-rich folds); (viii) protein motions (e.g. most mobile folds); and (ix) the level of similarity based on a comprehensive set of structural alignments (e.g. most structurally variable folds). The integration of whole-genome expression and protein-protein interaction data with structural information is a particularly novel feature of our system. We provide three ways of visualizing the rankings: a profiler emphasizing the progression of high and low ranks across many pre-selected attributes, a dynamic comparer for custom comparisons and a numerical rankings correlator. These allow one to directly compare very different attributes of a fold (e.g. expression level, genome occurrence and maximum motion) in the uniform numerical format of ranks. This uniform framework, in turn, highlights the way that the frequency of many of the attributes falls off with approximate power-law behavior (i.e. according to V(-b), for attribute value V and constant exponent b), with a few folds having large values and most having small values.
Figures
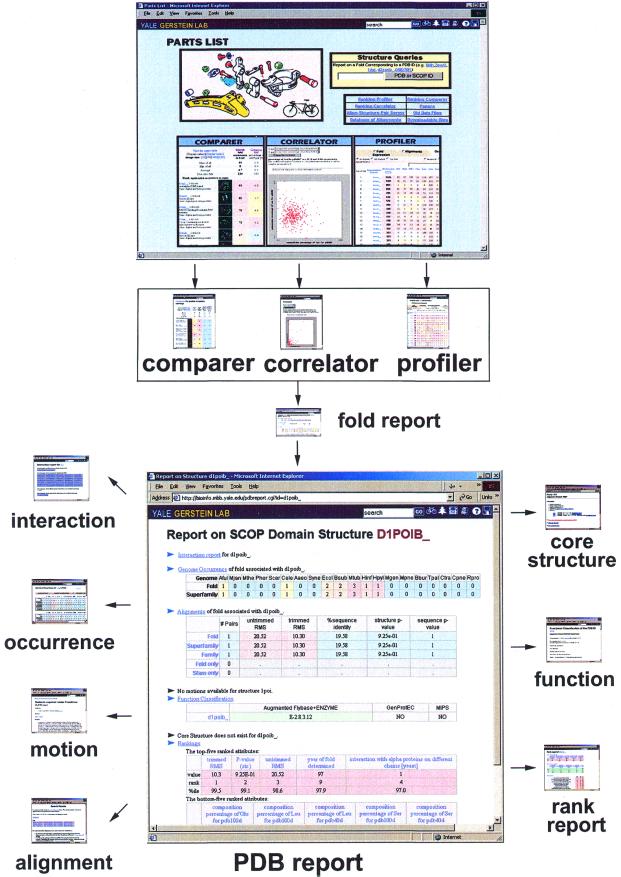
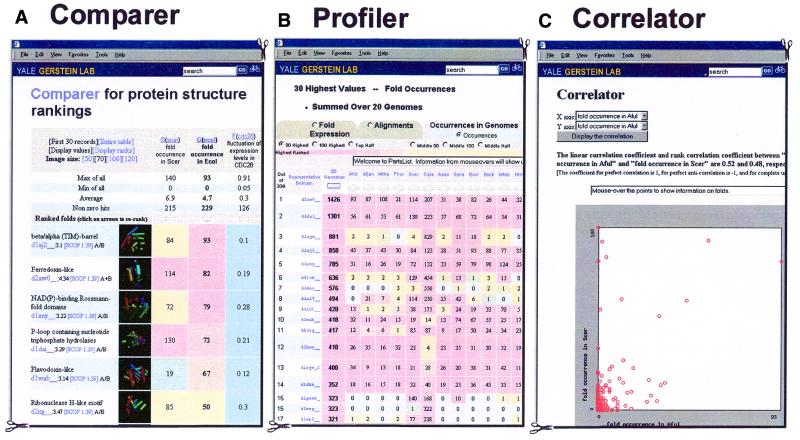
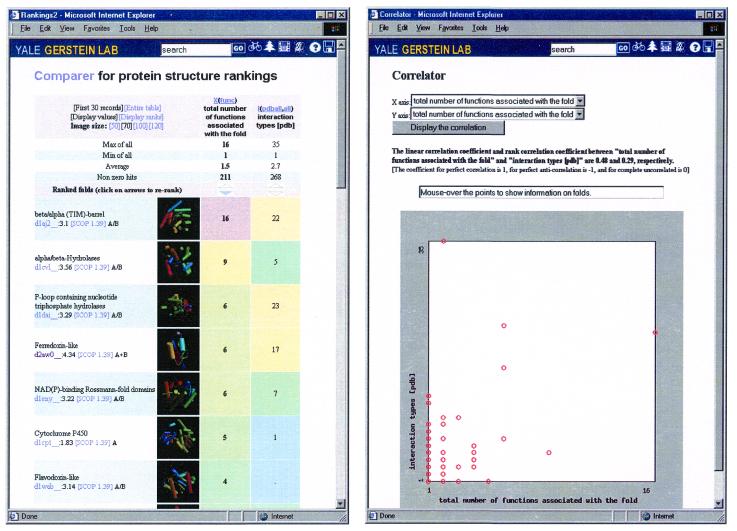
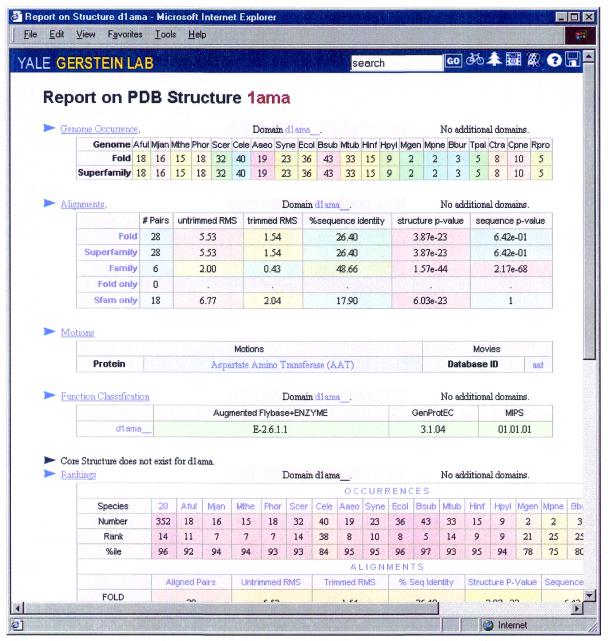
References
-
- Chothia C. (1992) Proteins. One thousand families for the molecular biologist. Nature, 357, 543–544. - PubMed
-
- Brenner S.E., Hubbard,T., Murzin,A. and Chothia,C. (1995) Gene duplications in H. influenzae. Nature, 378, 140. - PubMed
-
- Wolf Y.I., Grishin,N.V. and Koonin,E.V. (2000) Estimating the number of protein folds and families from complete genome data. J. Mol. Biol., 299, 897–905. - PubMed
-
- The C. elegans Sequencing Consortium (1998) Genome sequence of the nematode C. elegans: a platform for investigating biology. Science, 282, 2012–2018. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
