

A tandem repeats database for bacterial genomes: application to the genotyping of Yersinia pestis and Bacillus anthracis
- PMID: 11299044
- PMCID: PMC31411
- DOI: 10.1186/1471-2180-1-2
A tandem repeats database for bacterial genomes: application to the genotyping of Yersinia pestis and Bacillus anthracis
Abstract
Background: Some pathogenic bacteria are genetically very homogeneous, making strain discrimination difficult. In the last few years, tandem repeats have been increasingly recognized as markers of choice for genotyping a number of pathogens. The rapid evolution of these structures appears to contribute to the phenotypic flexibility of pathogens. The availability of whole-genome sequences has opened the way to the systematic evaluation of tandem repeats diversity and application to epidemiological studies.
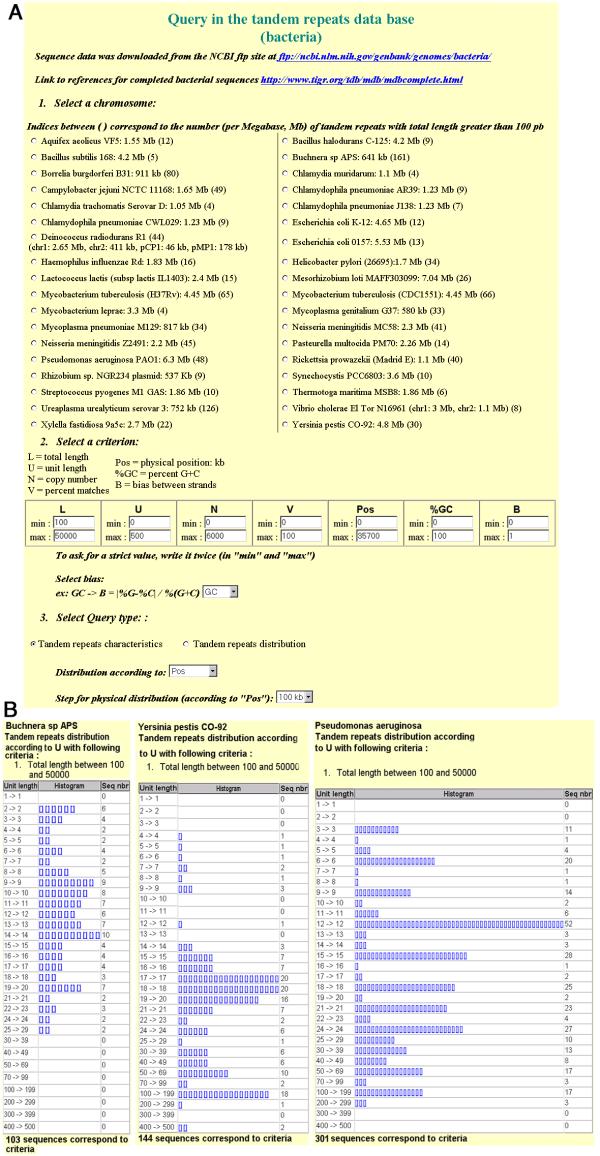
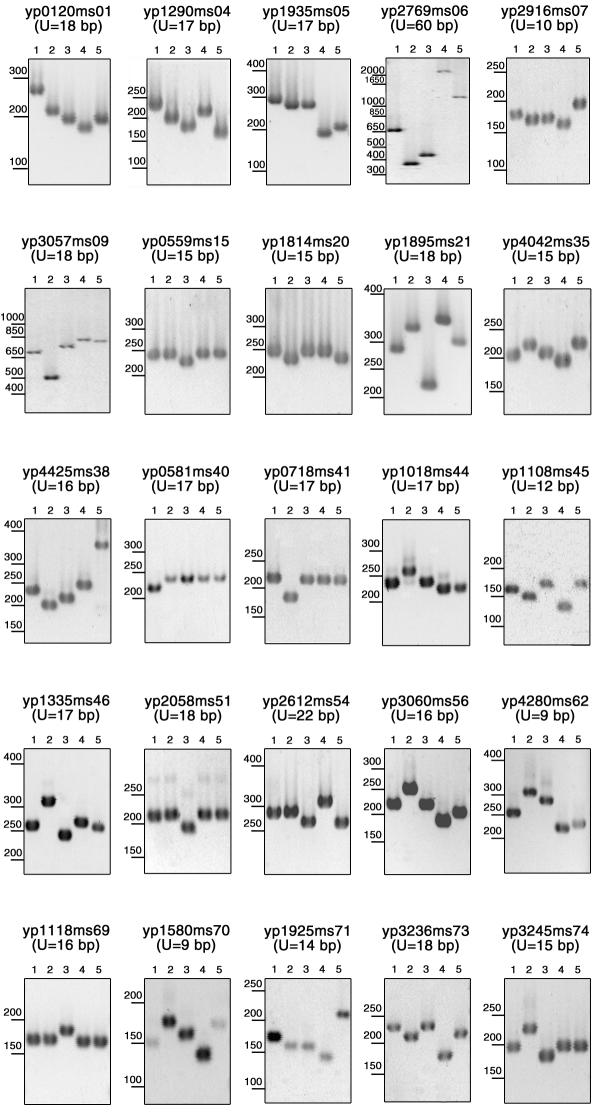
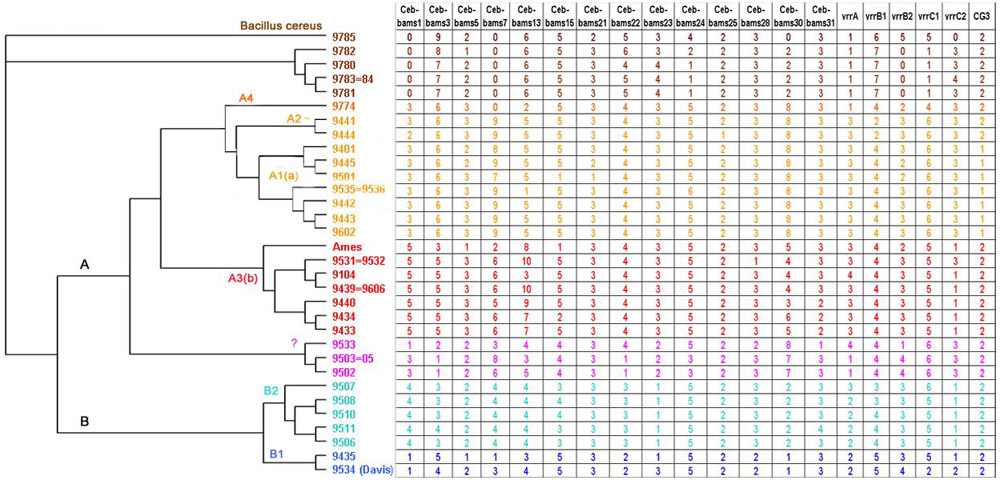
Results: This report presents a database (http://minisatellites.u-psud.fr) of tandem repeats from publicly available bacterial genomes which facilitates the identification and selection of tandem repeats. We illustrate the use of this database by the characterization of minisatellites from two important human pathogens, Yersinia pestis and Bacillus anthracis. In order to avoid simple sequence contingency loci which may be of limited value as epidemiological markers, and to provide genotyping tools amenable to ordinary agarose gel electrophoresis, only tandem repeats with repeat units at least 9 bp long were evaluated. Yersinia pestis contains 64 such minisatellites in which the unit is repeated at least 7 times. An additional collection of 12 loci with at least 6 units, and a high internal conservation were also evaluated. Forty-nine are polymorphic among five Yersinia strains (twenty-five among three Y. pestis strains). Bacillus anthracis contains 30 comparable structures in which the unit is repeated at least 10 times. Half of these tandem repeats show polymorphism among the strains tested.
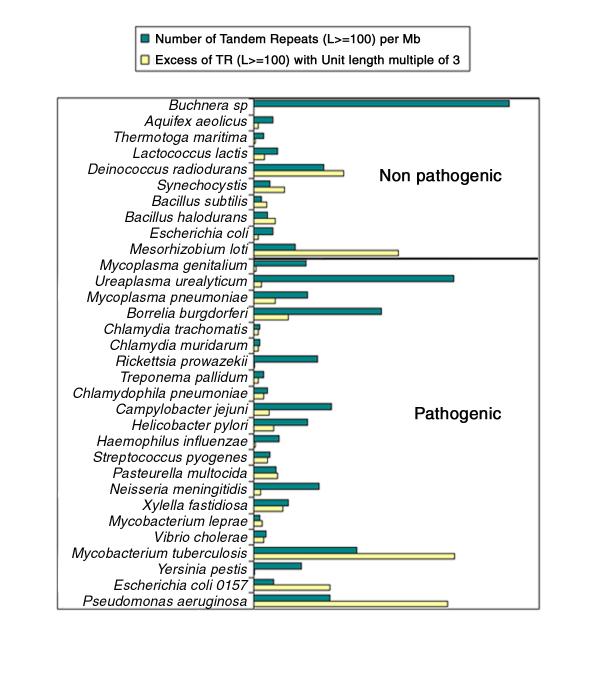
Conclusions: Analysis of the currently available bacterial genome sequences classifies Bacillus anthracis and Yersinia pestis as having an average (approximately 30 per Mb) density of tandem repeat arrays longer than 100 bp when compared to the other bacterial genomes analysed to date. In both cases, testing a fraction of these sequences for polymorphism was sufficient to quickly develop a set of more than fifteen informative markers, some of which show a very high degree of polymorphism. In one instance, the polymorphism information content index reaches 0.82 with allele length covering a wide size range (600-1950 bp), and nine alleles resolved in the small number of independent Bacillus anthracis strains typed here.
Figures

References
-
- Frothingham R, Meeker-O'Connell WA. Genetic diversity in the Mycobacterium tuberculosis complex based on variable numbers of tandem DNA repeats. Microbiology. 1998;144:1189–96. - PubMed
-
- Supply P, Mazars E, Lesjean S, Vincent V, Gicquel B, Locht C. Variable human minisatellite-like regions in the Mycobacterium tuberculosis genome. Mol Microbiol. 2000;36:762–71. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
