

MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin
- PMID: 11309679
- PMCID: PMC1226090
- DOI: 10.1086/320109
MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin
Abstract
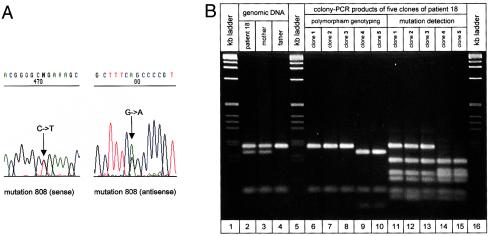
Rett syndrome (RTT) is an X-linked neurodevelopmental disorder that apparently is lethal in male embryos. RTT almost exclusively affects female offspring and, in 99.5% of all cases, is sporadic and due to de novo mutations in the MECP2 gene. Familial cases of RTT are rare and are due to X-chromosomal inheritance from a carrier mother. We analyzed the parental origin of MECP2 mutations in sporadic cases of RTT, by analysis of linkage between the mutation in the MECP2 gene and intronic polymorphisms in 27 families with 15 different mutations, and we found a high predominance of mutations of paternal origin in 26 of 27 cases (P<.001). The paternal origin was independent of type of mutation and was found for single-base exchanges as well as for deletions. Parents were not of especially advanced age. We conclude that de novo mutations in RTT occur almost exclusively on the paternally derived X chromosome and that this is most probably the cause for the high female:male ratio observed in patients with RTT. Affected males recently have been described in a few cases of familial inheritance. Identification of the parental origin may be useful to distinguish between the sporadic form of RTT and a potentially familial form. This distinction will allow geneticists to offer more-specific counseling and discriminate between higher (maternal origin) and lower (paternal origin) recurrence risk.
Figures
References
Electronic-Database Information
-
- dbSNP, http://www.ncbi.nlm.nih.gov/SNP/ (for intragenic SNPs, accession numbers rs760103, rs1474485, rs1042870, rs1042873, and rs1474486)
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for MECP2 [MIM 300005] and RTT [MIM 312750])
References
-
- Amano K, Nomura Y, Segawa M, Yamakawa K (2000) Mutational analysis of the MECP2 gene in Japanese patients with Rett syndrome. J Hum Genet 45:231–236 - PubMed
-
- Amir RE, Van den Veyver IB, Schultz R, Malicki DM, Tran CQ, Dahle EJ, Philippi A, Timar L, Percy AK, Motil KJ, Lichtarge O, Smith EO, Glaze DG, Zoghbi HY (2000) Influence of mutation type and X chromosome inactivation on Rett syndrome phenotypes. Ann Neurol 47:670–679 - PubMed
-
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23:185–188 - PubMed
-
- Bienvenu T, Carrie A, de Roux N, Vinet MC, Jonveaux P, Couvert P, Villard L, Arzimanoglou A, Beldjord C, Fontes M, Tardieu M, Chelly J (2000) MECP2 mutations account for most cases of typical forms of Rett syndrome. Hum Mol Genet 9:1377–1384 - PubMed
-
- Bohossian HB, Skaletsky H, Page DC (2000) Unexpectedly similar rates of nucleotide substitution found in male and female homonids. Nature 406:622–625 - PubMed
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
