

Analysis and characterization of the complete genome of tupaia (tree shrew) herpesvirus
- PMID: 11312357
- PMCID: PMC114240
- DOI: 10.1128/JVI.75.10.4854-4870.2001
Analysis and characterization of the complete genome of tupaia (tree shrew) herpesvirus
Abstract
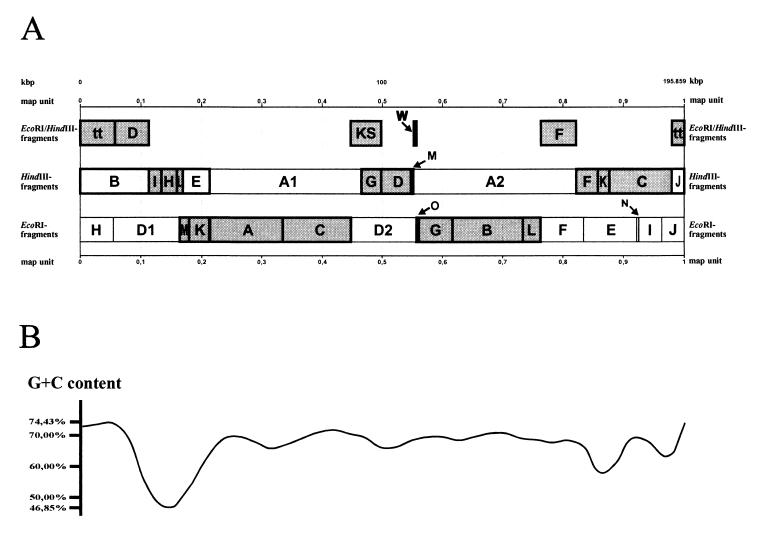
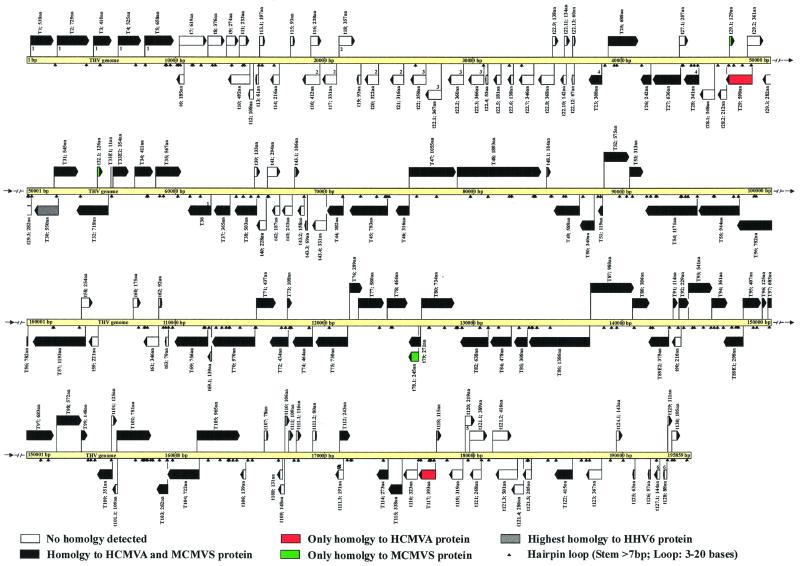
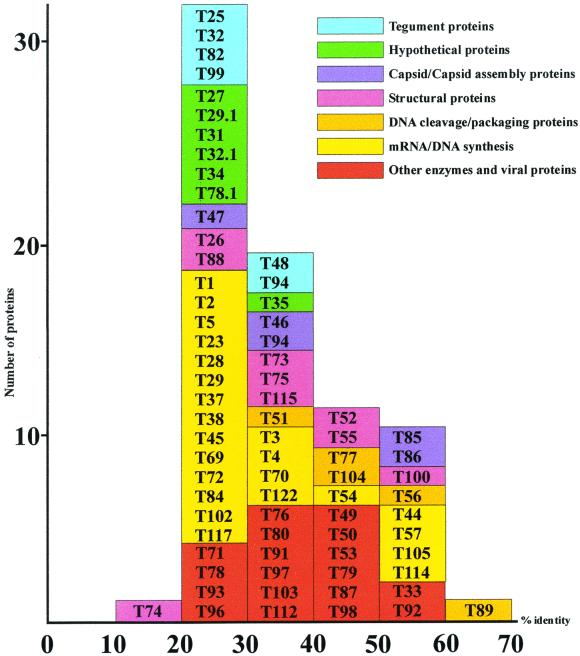
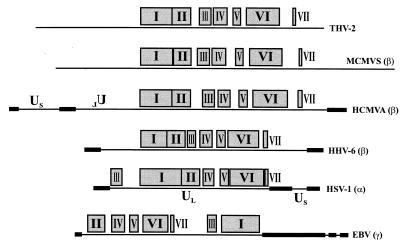
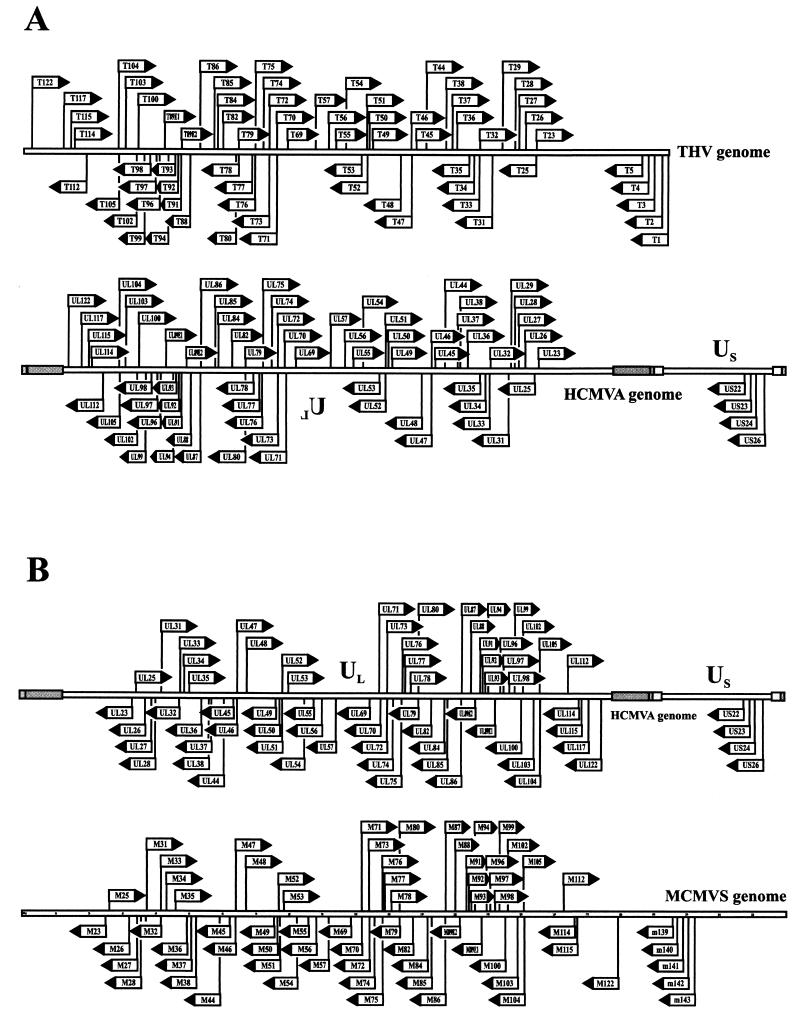
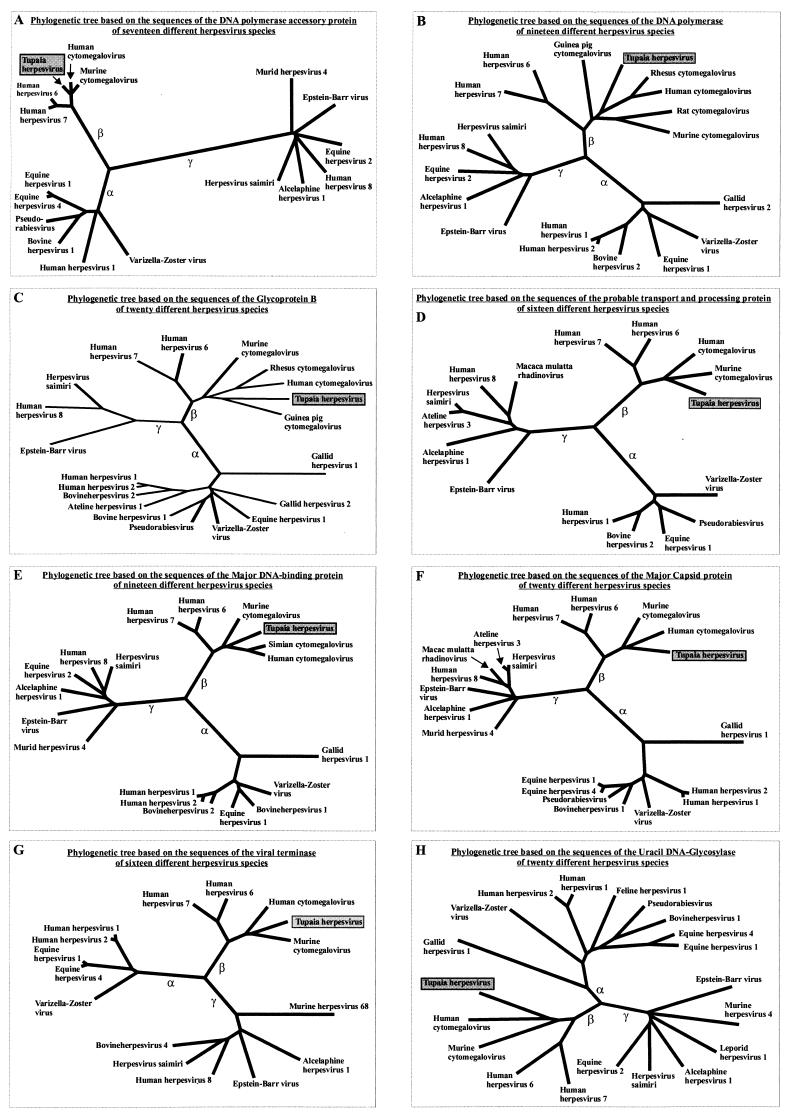
The tupaia herpesvirus (THV) was isolated from spontaneously degenerating tissue cultures of malignant lymphoma, lung, and spleen cell cultures of tree shrews (Tupaia spp.). The determination of the complete nucleotide sequence of the THV strain 2 genome resulted in a 195,857-bp-long, linear DNA molecule with a G+C content of 66.5%. The terminal regions of the THV genome and the loci of conserved viral genes were found to be G+C richer. Furthermore, no large repetitive DNA sequences could be identified. This is in agreement with the previous classification of THV as the prototype species of herpesvirus genome group F. The search for potential coding regions resulted in the identification of 158 open reading frames (ORFs) regularly distributed on both DNA strands. Seventy-six out of the 158 ORFs code for proteins that are significantly homologous to known herpesvirus proteins. The highest homologies found were to primate and rodent cytomegaloviruses. Biological properties, protein homologies, the arrangement of conserved viral genes, and phylogenetic analysis revealed that THV is a member of the subfamily Betaherpesvirinae. The evolutionary lineages of THV and the cytomegaloviruses seem to have branched off from a common ancestor. In addition, it was found that the arrangements of conserved genes of THV and murine cytomegalovirus strain Smith, both of which are not able to form genomic isomers, are colinear with two different human cytomegalovirus (HCMV) strain AD169 genomic isomers that differ from each other in the orientation of the long unique region. The biological properties and the high degree of relatedness of THV to the mammalian cytomegaloviruses allow the consideration of THV as a model system for investigation of HCMV pathogenicity.
Figures
References
-
- Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
-
- Baer R, Bankier A T, Biggin M D, Deininger P L, Farrell P J, Gibson T J, Hatfull G, Hudson G S, Satchwell S C, Seguin C, Tuffnell P S, Barrell B G. DNA sequence and expression of the B95–8 Epstein-Barr virus genome. Nature. 1984;310:207–211. - PubMed
-
- Bahr U, Springfeld C, Tidona C A, Darai G. Structural organization of a conserved gene cluster of Tupaia herpesvirus encoding the DNA polymerase, glycoprotein B, a probable processing and transport protein, and the major DNA binding protein. Virus Res. 1999;60:123–136. - PubMed
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Miscellaneous
