

Functional alterations in gap junction channels formed by mutant forms of connexin 32: evidence for loss of function as a pathogenic mechanism in the X-linked form of Charcot-Marie-Tooth disease
- PMID: 11325342
- PMCID: PMC4517190
- DOI: 10.1016/s0006-8993(00)03327-8
Functional alterations in gap junction channels formed by mutant forms of connexin 32: evidence for loss of function as a pathogenic mechanism in the X-linked form of Charcot-Marie-Tooth disease
Abstract
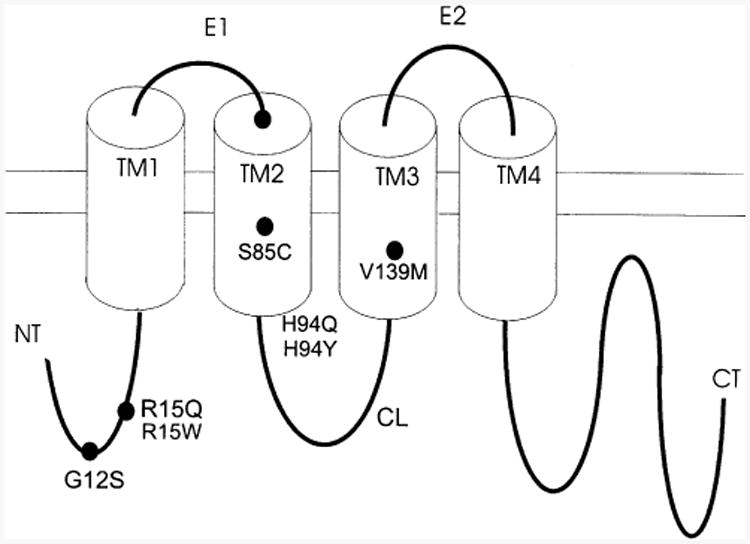
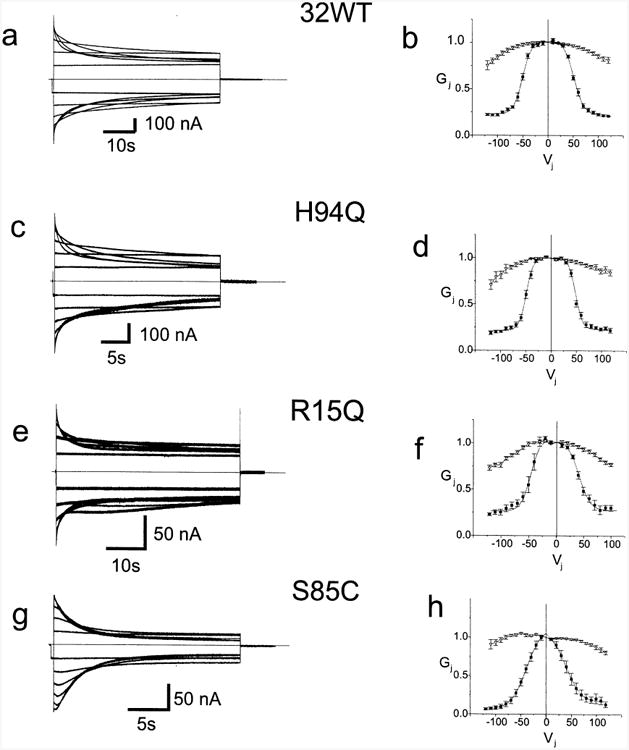
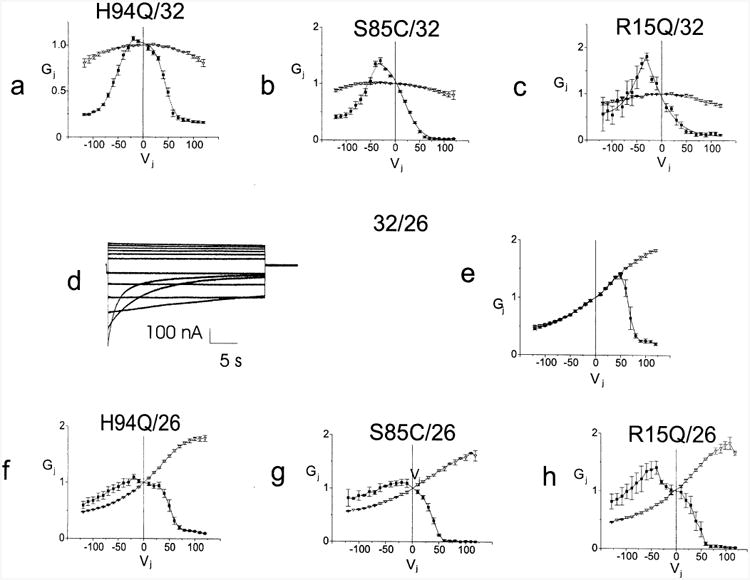
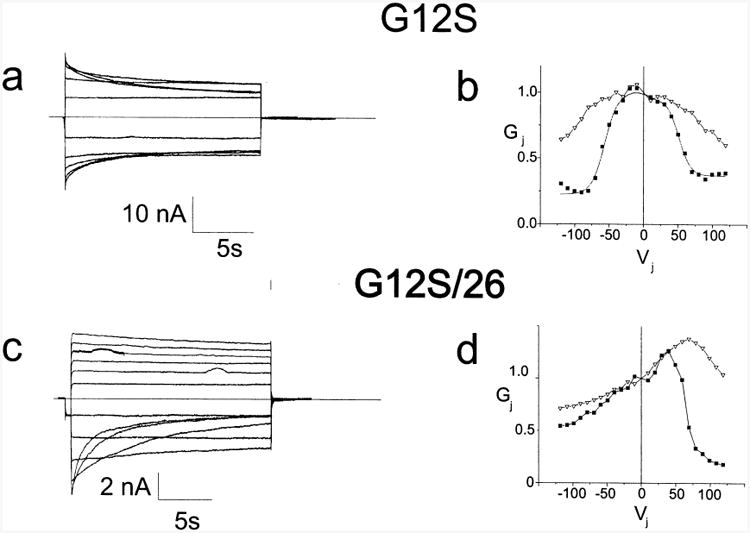
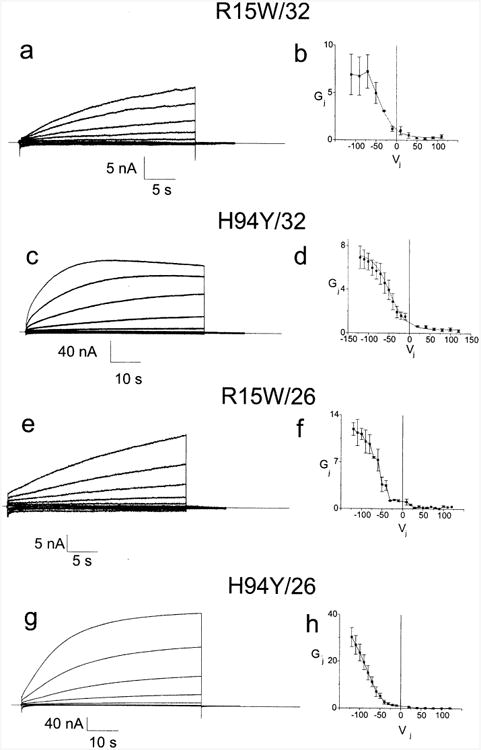
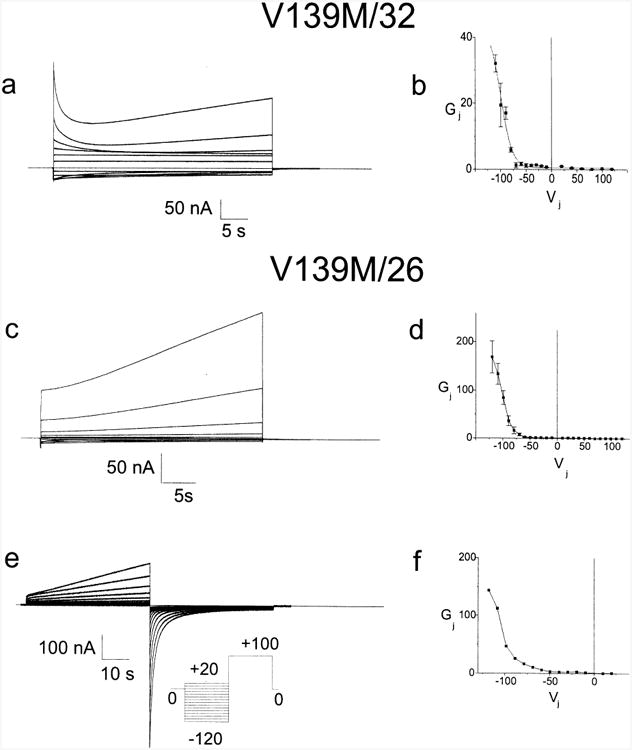
CMTX, the X-linked form of Charcot-Marie-Tooth disease, is an inherited peripheral neuropathy arising in patients with mutations in the gene encoding the gap junction protein connexin 32 (Cx32). In this communication, we describe the expression levels and biophysical parameters of seven mutant forms of Cx32 associated with CMTX, when expressed in paired Xenopus oocytes. Paired oocytes expressing the R15Q and H94Q mutants show junctional conductances not statistically different from that determined for Cx32WT, though both show a trend toward reduced levels. The S85C and G12S mutants induce reduced levels of junctional conductance. Three other mutants (R15W, H94Y and V139M) induce no conductance above baseline when expressed in paired oocytes. Analysis of the conductance voltage relations for these mutants shows that the reduced levels of conductance are entirely (H94Y and V139M) or partly (S85C and R15W) explicable by a reduced open probability of the mutant hemichannels. The R15Q and H94Q mutations also show alterations in the conductance voltage relations that would be expected to minimally (H94Q) or moderately (R15Q) reduce the available gap junction communication pathway. The reduction in G12S induced conductance cannot be explained by alterations in hemichannel open probability and are more likely due to reduced junction formation. These results demonstrate that many CMTX mutations lead to loss of function of Cx32. For these mutations, the loss of function model is likely to explain the pathogenesis of CMTX.
Figures
References
-
- Abel A, Bone LJ, Messing A, Scherer SS, Fischbeck KH. Studies in transgenic mice indicate a loss of connexin32 function in X-linked Charcot-Marie-Tooth disease. J Neuropathol Exp Neurol. 1999;58:702–710. - PubMed
-
- Ainsworth PJ, Bolton CF, Murphy BC, Stuart JA, Hahn AF. Genotype/phenotype correlation in affected individuals of a family with a deletion of the entire coding sequence of the connexin 32 gene. Hum Genet. 1998;103:242–244. - PubMed
-
- Bergoffen J, Scherer SS, Wang S, Scott MO, Bone LJ, Paul DL, Chen K, Lensch MW, Chance PF, Fischbeck KH. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science. 1993;262:2039–2042. - PubMed
-
- Bevans CG, Kordel M, Rhee SK, Harris AL. Isoform composition of connexin channels determines selectivity among second messengers and uncharged molecules. J Biol Chem. 1998;273:2808–2816. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
