

Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis
- PMID: 11331305
- PMCID: PMC2190573
- DOI: 10.1083/jcb.153.3.543
Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis
Abstract
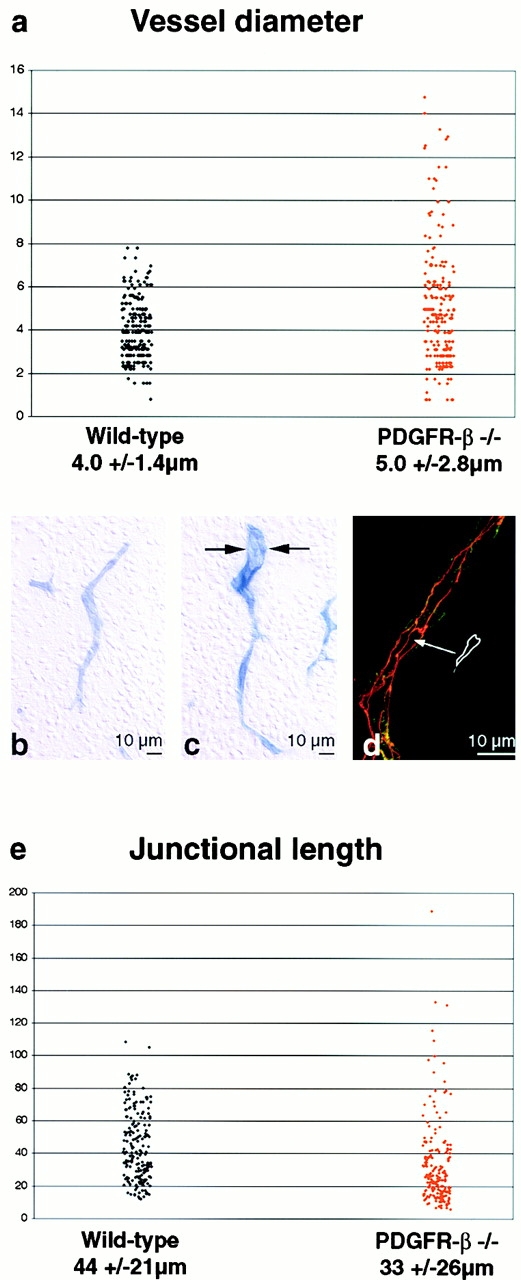
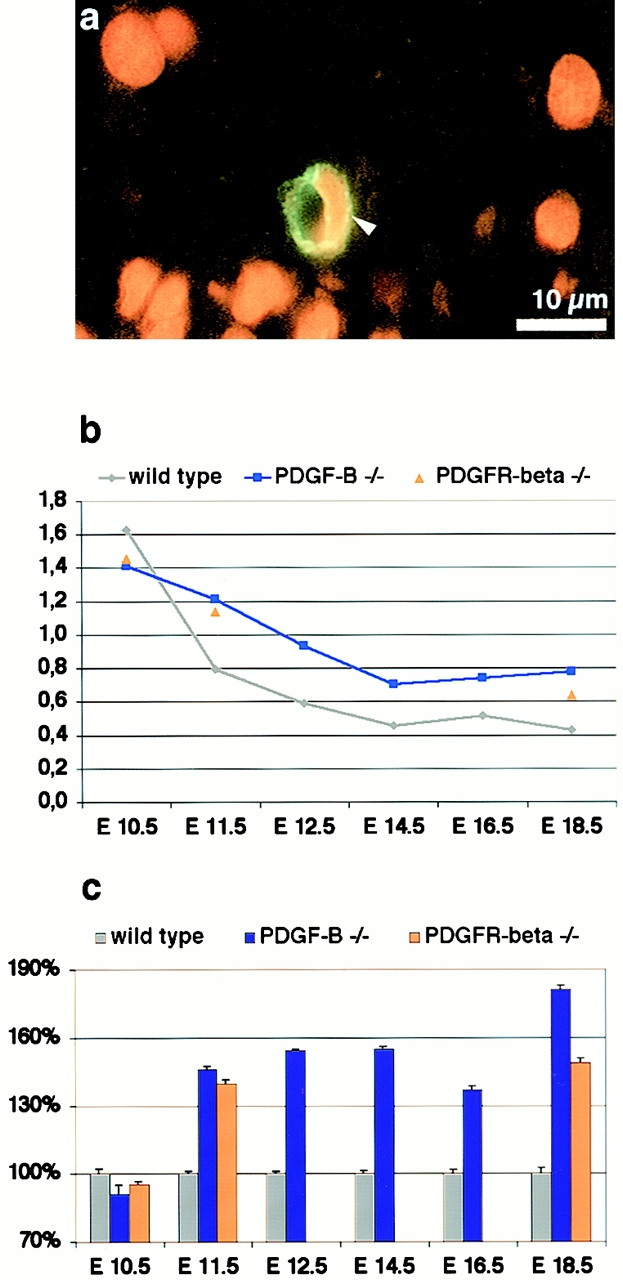
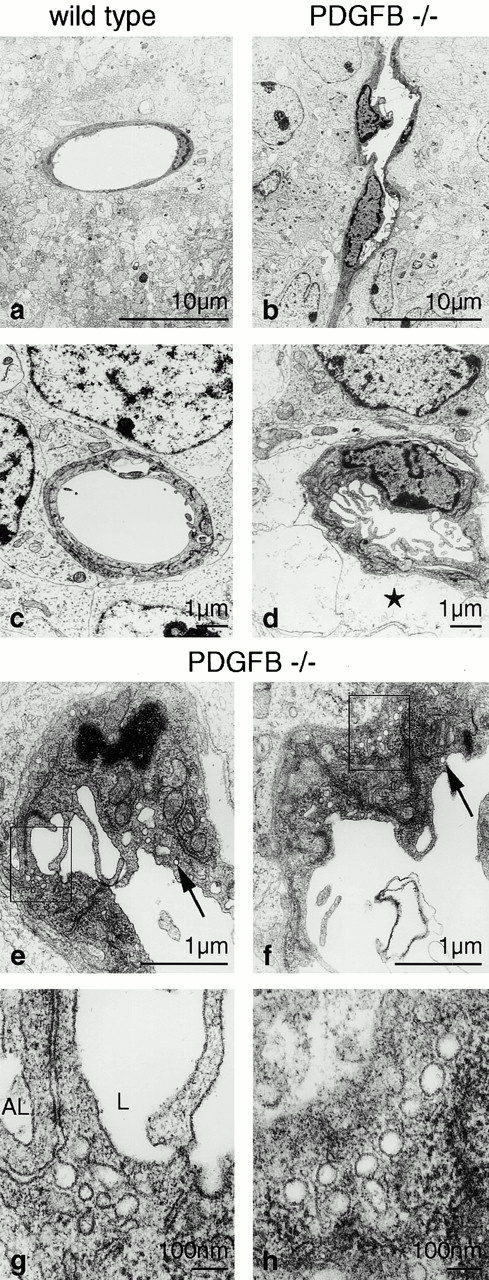
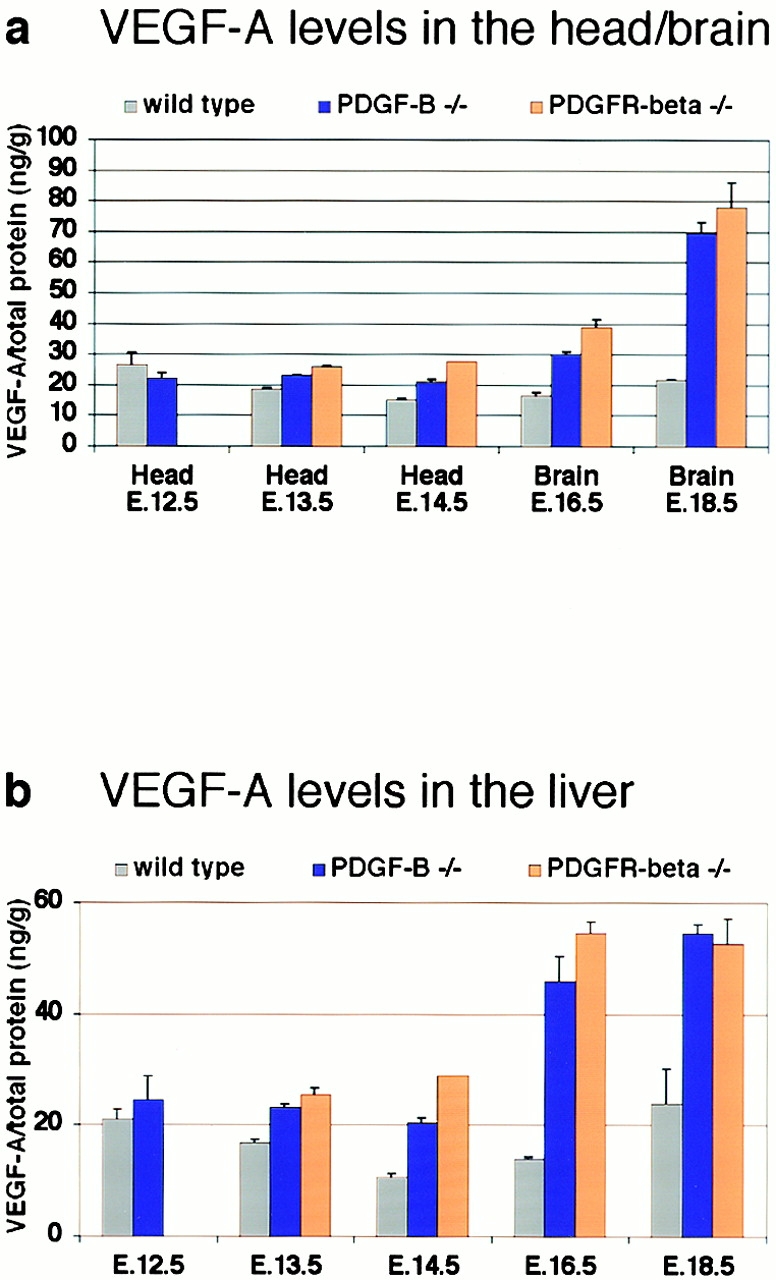
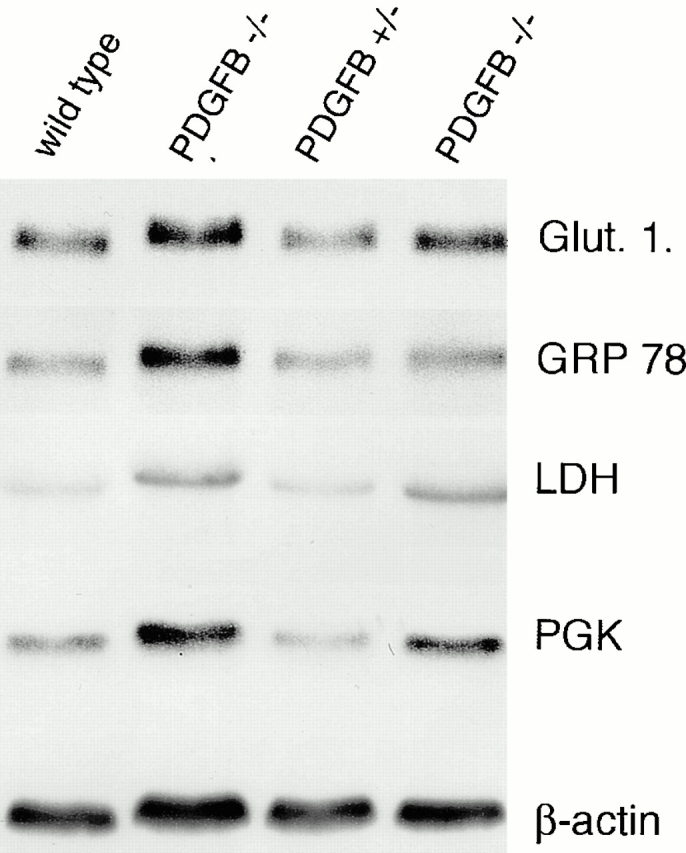
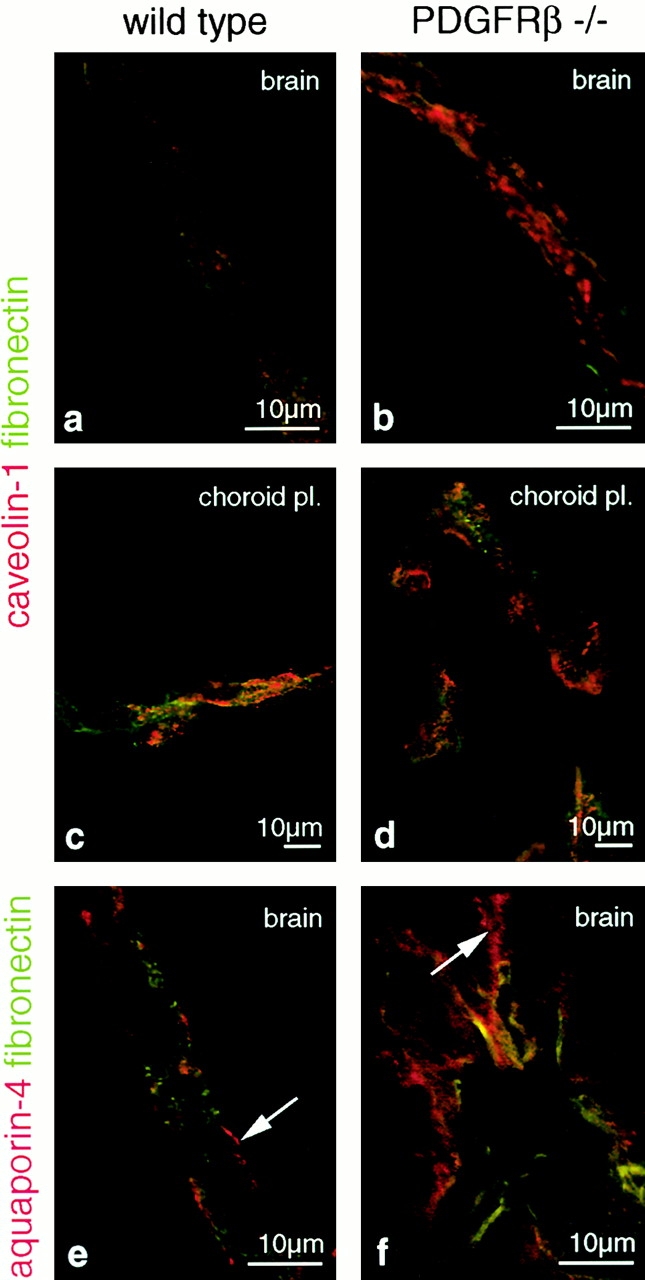
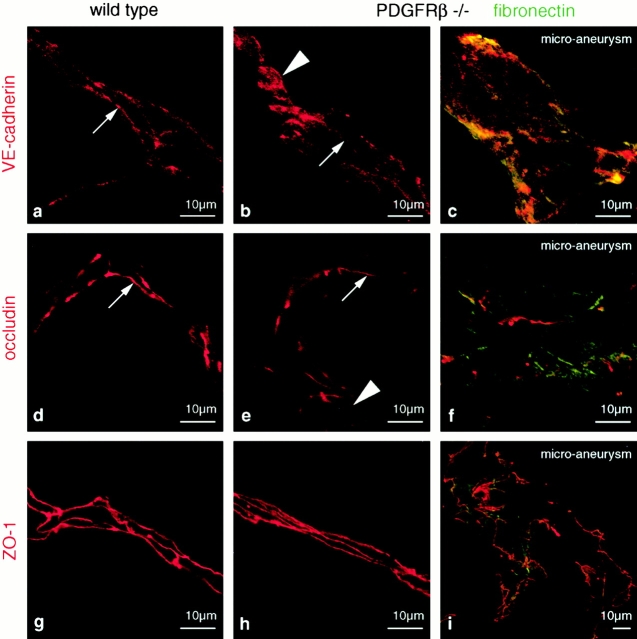
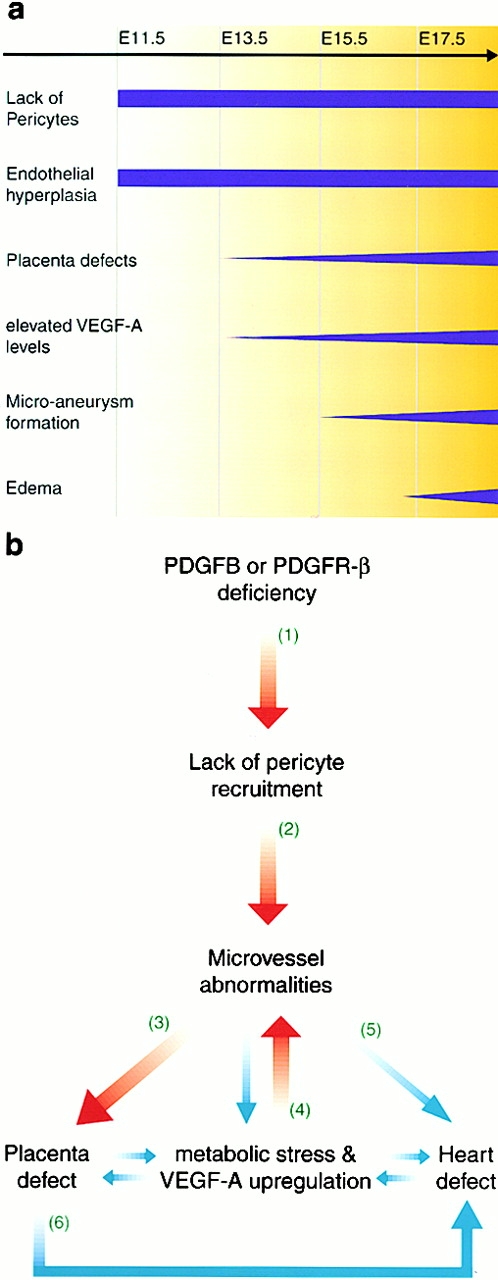
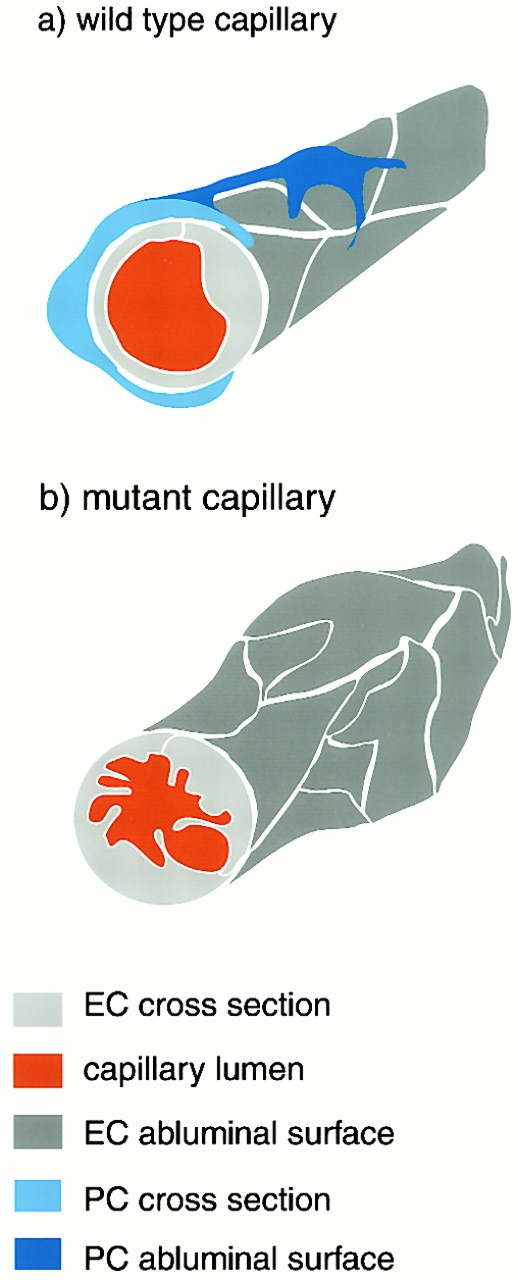
The association of pericytes (PCs) to newly formed blood vessels has been suggested to regulate endothelial cell (EC) proliferation, survival, migration, differentiation, and vascular branching. Here, we addressed these issues using PDGF-B-- and PDGF receptor-beta (PDGFR-beta)--deficient mice as in vivo models of brain angiogenesis in the absence of PCs. Quantitative morphological analysis showed that these mutants have normal microvessel density, length, and number of branch points. However, absence of PCs correlates with endothelial hyperplasia, increased capillary diameter, abnormal EC shape and ultrastructure, changed cellular distribution of certain junctional proteins, and morphological signs of increased transendothelial permeability. Brain endothelial hyperplasia was observed already at embryonic day (E) 11.5 and persisted throughout development. From E 13.5, vascular endothelial growth factor-A (VEGF-A) and other genes responsive to metabolic stress became upregulated, suggesting that the abnormal microvessel architecture has systemic metabolic consequences. VEGF-A upregulation correlated temporally with the occurrence of vascular abnormalities in the placenta and dilation of the heart. Thus, although PC deficiency appears to have direct effects on EC number before E 13.5, the subsequent increased VEGF-A levels may further abrogate microvessel architecture, promote vascular permeability, and contribute to formation of the edematous phenotype observed in late gestation PDGF-B and PDGFR-beta knock out embryos.
Figures

References
-
- Barak Y., Nelson M.C., Ong E.S., Jones Y.Z., Ruiz-Lozano P., Chien K.R., Koder A., Evans R.M. PPAR γ is required for placental, cardiac, and adipose tissue development. Mol. Cell. 1999;4:585–595. - PubMed
-
- Beck L.J., D'Amore P.A. Vascular developmentcellular and molecular regulation. FASEB J. 1997;11:365–373. - PubMed
-
- Benjamin L., Hemo I., Keshet E. A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development. 1998;125:1591–1598. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
