

Nemaline myopathy caused by mutations in the muscle alpha-skeletal-actin gene
- PMID: 11333380
- PMCID: PMC1226120
- DOI: 10.1086/320605
Nemaline myopathy caused by mutations in the muscle alpha-skeletal-actin gene
Abstract
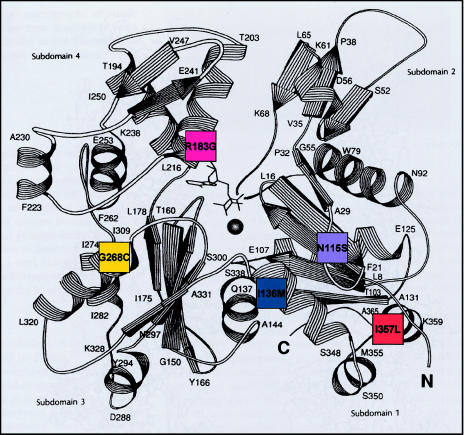
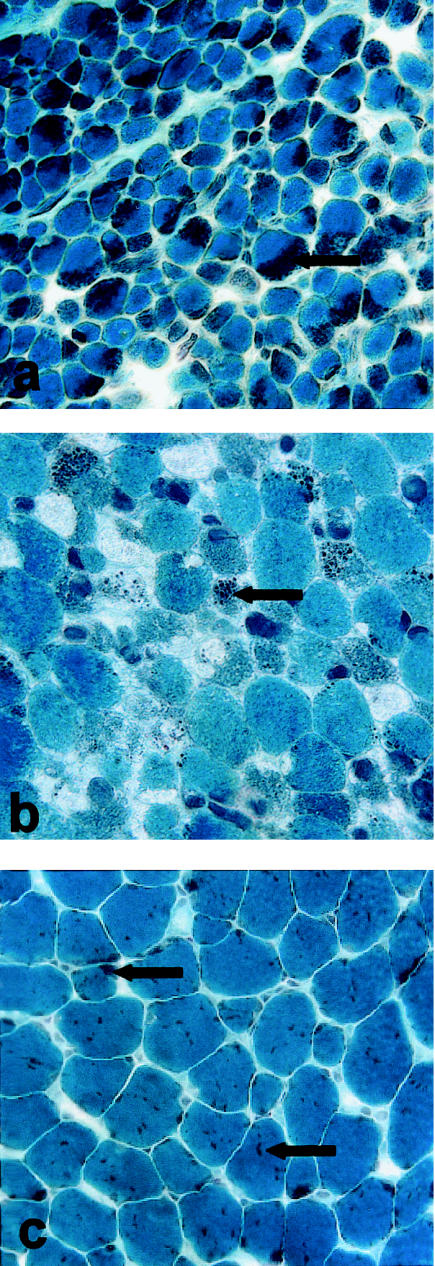
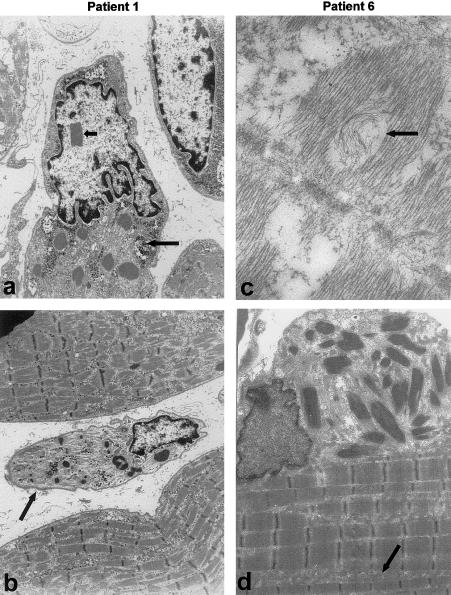
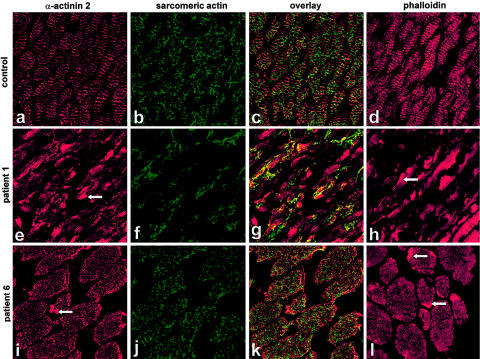
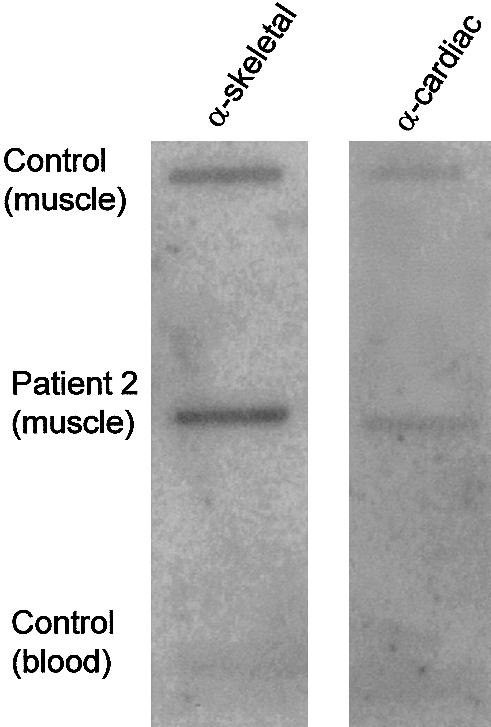
Nemaline myopathy (NM) is a clinically and genetically heterogeneous disorder characterized by muscle weakness and the presence of nemaline bodies (rods) in skeletal muscle. Disease-causing mutations have been reported in five genes, each encoding a protein component of the sarcomeric thin filament. Recently, we identified mutations in the muscle alpha-skeletal-actin gene (ACTA1) in a subset of patients with NM. In the present study, we evaluated a new series of 35 patients with NM. We identified five novel missense mutations in ACTA1, which suggested that mutations in muscle alpha-skeletal actin account for the disease in approximately 15% of patients with NM. The mutations appeared de novo and represent new dominant mutations. One proband subsequently had two affected children, a result consistent with autosomal dominant transmission. The seven patients exhibited marked clinical variability, ranging from severe congenital-onset weakness, with death from respiratory failure during the 1st year of life, to a mild childhood-onset myopathy, with survival into adulthood. There was marked variation in both age at onset and clinical severity in the three affected members of one family. Common pathological features included abnormal fiber type differentiation, glycogen accumulation, myofibrillar disruption, and "whorling" of actin thin filaments. The percentage of fibers with rods did not correlate with clinical severity; however, the severe, lethal phenotype was associated with both severe, generalized disorganization of sarcomeric structure and abnormal localization of sarcomeric actin. The marked variability, in clinical phenotype, among patients with different mutations in ACTA1 suggests that both the site of the mutation and the nature of the amino acid change have differential effects on thin-filament formation and protein-protein interactions. The intrafamilial variability suggests that alpha-actin genotype is not the sole determinant of phenotype.
Figures

Similar articles
-
Mild phenotype of nemaline myopathy with sleep hypoventilation due to a mutation in the skeletal muscle alpha-actin (ACTA1) gene.Neuromuscul Disord. 2001 Jan;11(1):35-40. doi: 10.1016/s0960-8966(00)00167-x. Neuromuscul Disord. 2001. PMID: 11166164
-
Heterogeneity of nemaline myopathy cases with skeletal muscle alpha-actin gene mutations.Ann Neurol. 2004 Jul;56(1):86-96. doi: 10.1002/ana.20157. Ann Neurol. 2004. PMID: 15236405
-
Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and nemaline myopathy.Nat Genet. 1999 Oct;23(2):208-12. doi: 10.1038/13837. Nat Genet. 1999. PMID: 10508519
-
Clinical and Histologic Findings in ACTA1-Related Nemaline Myopathy: Case Series and Review of the Literature.Pediatr Neurol. 2017 Oct;75:11-16. doi: 10.1016/j.pediatrneurol.2017.04.002. Epub 2017 Apr 7. Pediatr Neurol. 2017. PMID: 28780987 Review.
-
Skeletal muscle α-actin diseases (actinopathies): pathology and mechanisms.Acta Neuropathol. 2013 Jan;125(1):19-32. doi: 10.1007/s00401-012-1019-z. Epub 2012 Jul 24. Acta Neuropathol. 2013. PMID: 22825594 Review.
Cited by
-
Cofilin Loss in Drosophila Muscles Contributes to Muscle Weakness through Defective Sarcomerogenesis during Muscle Growth.Cell Rep. 2020 Jul 21;32(3):107893. doi: 10.1016/j.celrep.2020.107893. Cell Rep. 2020. PMID: 32697999 Free PMC article.
-
A rare structural myopathy: Nemaline myopathy.Turk Pediatri Ars. 2019 Mar 1;54(1):49-52. doi: 10.5152/TurkPediatriArs.2018.4402. eCollection 2019. Turk Pediatri Ars. 2019. PMID: 31217710 Free PMC article.
-
Pathogenic variants in TNNC2 cause congenital myopathy due to an impaired force response to calcium.J Clin Invest. 2021 May 3;131(9):e145700. doi: 10.1172/JCI145700. J Clin Invest. 2021. PMID: 33755597 Free PMC article.
-
Phenotypes induced by NM causing alpha-skeletal muscle actin mutants in fibroblasts, Sol 8 myoblasts and myotubes.BMC Res Notes. 2009 Mar 10;2:40. doi: 10.1186/1756-0500-2-40. BMC Res Notes. 2009. PMID: 19284548 Free PMC article.
-
Absence of the Drosophila jump muscle actin Act79B is compensated by up-regulation of Act88F.Dev Dyn. 2018 Apr;247(4):642-649. doi: 10.1002/dvdy.24616. Epub 2018 Feb 6. Dev Dyn. 2018. PMID: 29318731 Free PMC article.
References
Electronic-Database Information
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for ACTA1 [MIM 102610])
References
-
- Adelstein RS, Eisenberg E (1980) Regulation and kinetics of the actin-myosin-ATP interaction. Annu Rev Biochem 49:921–956 - PubMed
-
- Blanchard A, Ohanian V, Critchley D (1989) The structure and function of α-actinin. J Muscle Res Cell Motil 10:280–289 - PubMed
-
- Collins T, Joya JE, Arkell RM, Ferguson V, Hardeman EC (1997) Reappearance of the minor α-sarcomeric actins in postnatal muscle. Am J Physiol 273:C1801–C1810 - PubMed
-
- Donner K, Ollikainen M, Pelin K, Gronholm M, Carpen O, Wallgren-Pettersson C, Ridanpaa M (2000) Mutations in the β-tropomyosin (TPM2) gene in rare cases of autosomal dominant nemaline myopathy. World Muscle Society abstract. Neuromuscul Disord 10:342–343
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
