

Niemann-Pick C1 disease: correlations between NPC1 mutations, levels of NPC1 protein, and phenotypes emphasize the functional significance of the putative sterol-sensing domain and of the cysteine-rich luminal loop
- PMID: 11333381
- PMCID: PMC1226124
- DOI: 10.1086/320606
Niemann-Pick C1 disease: correlations between NPC1 mutations, levels of NPC1 protein, and phenotypes emphasize the functional significance of the putative sterol-sensing domain and of the cysteine-rich luminal loop
Abstract
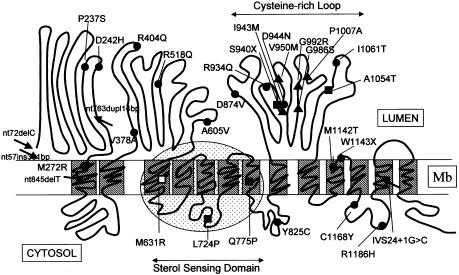
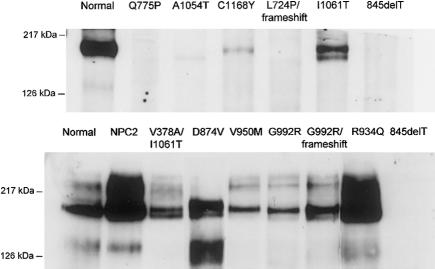
To obtain more information of the functional domains of the NPC1 protein, the mutational spectrum and the level of immunoreactive protein were investigated in skin fibroblasts from 30 unrelated patients with Niemann-Pick C1 disease. Nine of them were characterized by mild alterations of cellular cholesterol transport (the "variant" biochemical phenotype). The mutations showed a wide distribution to nearly all NPC1 domains, with a cluster (11/32) in a conserved NPC1 cysteine-rich luminal loop. Homozygous mutations in 14 patients and a phenotypically defined allele, combined with a new mutation, in a further 10 patients allowed genotype/phenotype correlations. Premature-termination-codon mutations, the three missense mutations in the sterol-sensing domain (SSD), and A1054T in the cysteine-rich luminal loop all occurred in patients with infantile neurological onset and "classic" (severe) cholesterol-trafficking alterations. By western blot, NPC1 protein was undetectable in the SSD missense mutations studied (L724P and Q775P) and essentially was absent in the A1054T missense allele. Our results thus enhance the functional significance of the SSD and demonstrate a correlation between the absence of NPC1 protein and the most severe neurological form. In the remaining missense mutations studied, corresponding to other disease presentations (including two adults with nonneurological disease), NPC1 protein was present in significant amounts of normal size, without clear-cut correlation with either the clinical phenotype or the "classic"/"variant" biochemical phenotype. Missense mutations in the cysteine-rich luminal loop resulted in a wide array of clinical and biochemical phenotypes. Remarkably, all five mutant alleles (I943M, V950M, G986S, G992R, and the recurrent P1007A) definitively correlated with the "variant" phenotype clustered within this loop, providing new insight on the functional complexity of the latter domain.
Figures
References
Electronic-Database Information
-
- Genbank, http://www.ncbi.nlm.nih.gov/Genbank/index.html (for NPC1 cDNA [accession number AF002020])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for Niemann-Pick C [MIM 257220]
References
-
- Baumkötter J, Freisinger P, Schneider KTM, Harzer K, Vanier MT, Pontz BF (1998) Fetal ascites: a rare presentation of Niemann-Pick disease type C. J Inher Metab Dis Suppl 21:118
-
- Blanchette-Mackie EJ (2000) Intracellular cholesterol trafficking: role of the NPC1 protein. Biochim Biophys Acta 1486:171–183 - PubMed
-
- Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, Gu J, et al (1997) Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 277:228–231 - PubMed
-
- Christomanou H, Vanier MT, Santambrogio P, Arosio P, Kleijer WJ, Harzer K (2000) Deficient ferritin immunoreactivity in tissues from Niemann-Pick type C patients: extension of findings to fetal tissues, H and L ferritin isoforms, but also one case of the rare Niemann-Pick C2 complementation group. Mol Genet Metab 70:196–202 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
