

The basic helix-loop-helix protein family: comparative genomics and phylogenetic analysis
- PMID: 11337472
- PMCID: PMC311049
- DOI: 10.1101/gr.177001
The basic helix-loop-helix protein family: comparative genomics and phylogenetic analysis
Abstract
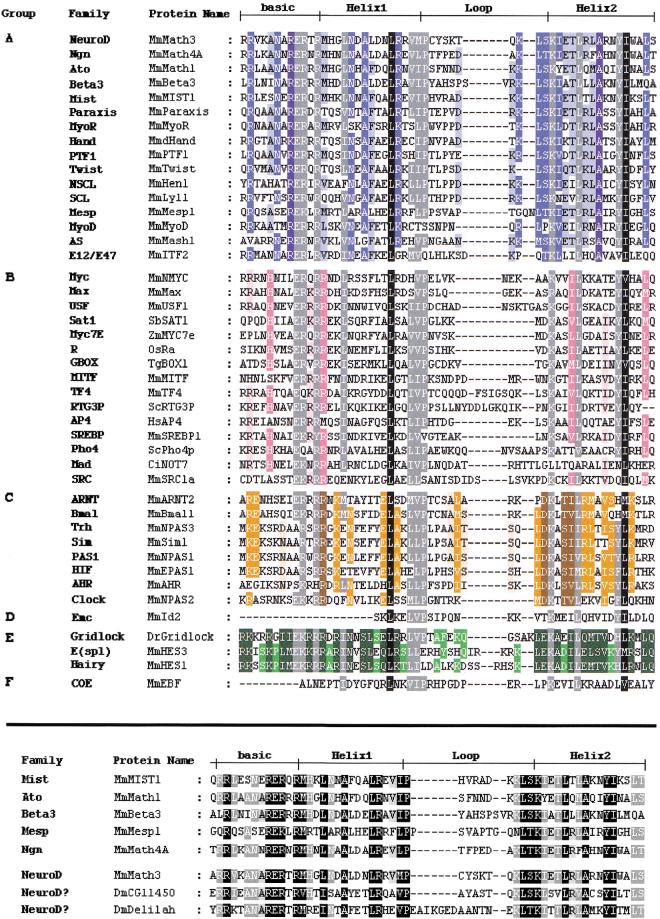
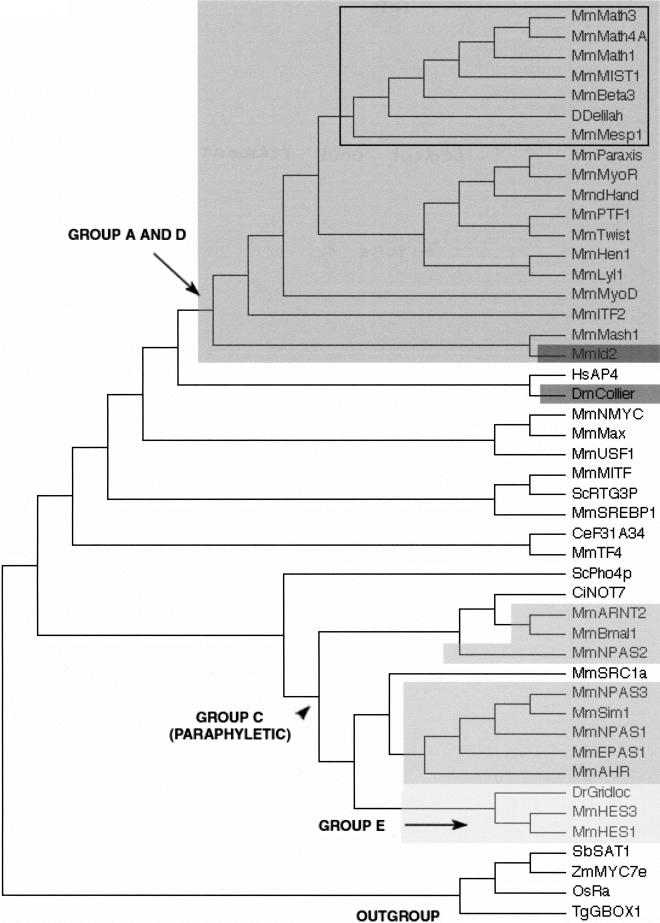
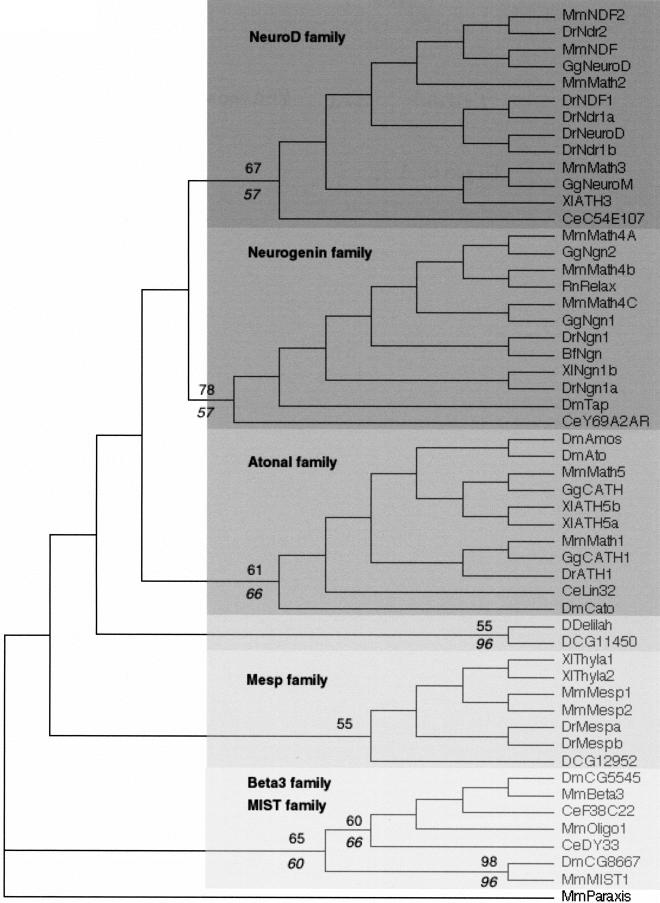
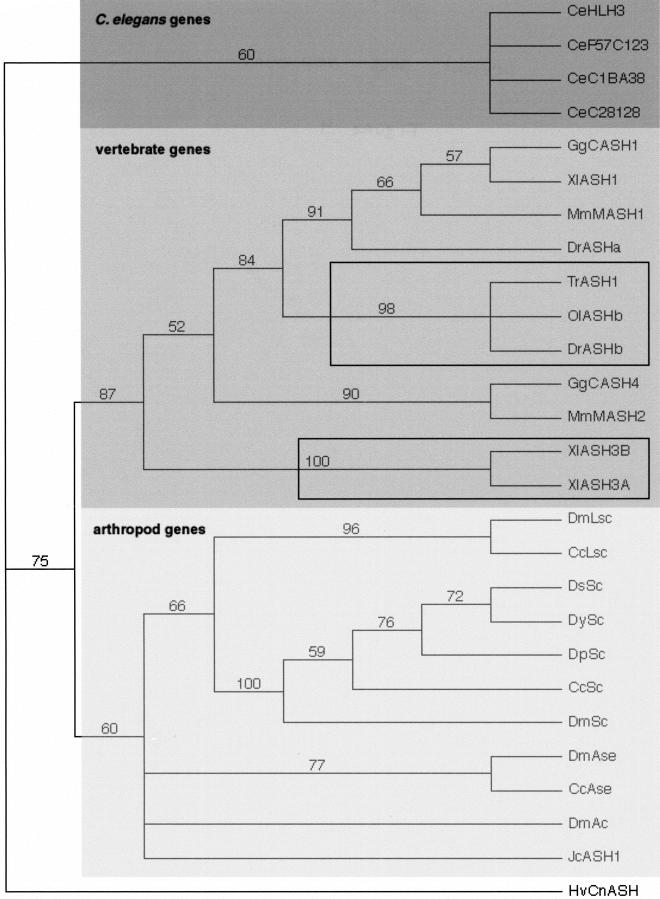
The basic Helix-Loop-Helix (bHLH) proteins are transcription factors that play important roles during the development of various metazoans including fly, nematode, and vertebrates. They are also involved in human diseases, particularly in cancerogenesis. We made an extensive search for bHLH sequences in the completely sequenced genomes of Caenorhabditis elegans and of Drosophila melanogaster. We found 35 and 56 different genes, respectively, which may represent the complete set of bHLH of these organisms. A phylogenetic analysis of these genes, together with a large number (>350) of bHLH from other sources, led us to define 44 orthologous families among which 36 include bHLH from animals only, and two have representatives in both yeasts and animals. In addition, we identified two bHLH motifs present only in yeast, and four that are present only in plants; however, the latter number is certainly an underestimate. Most animal families (35/38) comprise fly, nematode, and vertebrate genes, suggesting that their common ancestor, which lived in pre-Cambrian times (600 million years ago) already owned as many as 35 different bHLH genes.
Figures
References
-
- Adams MD, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. - PubMed
-
- Aguinaldo AMA, Turbeville JM, Linford LS, Rivera MC, Garey JR, Raff RA, Lake JA. Evidence for a clade of nematodes, arthropods and other moulting animals. Nature. 1997;387:489–493. - PubMed
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
-
- Arendt D, Nübler-Jung K. Comparisons of early nerve cord development in insects and vertebrates. Development. 1999;126:2309–2325. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases