

Computational inference of homologous gene structures in the human genome
- PMID: 11337476
- PMCID: PMC311055
- DOI: 10.1101/gr.175701
Computational inference of homologous gene structures in the human genome
Abstract
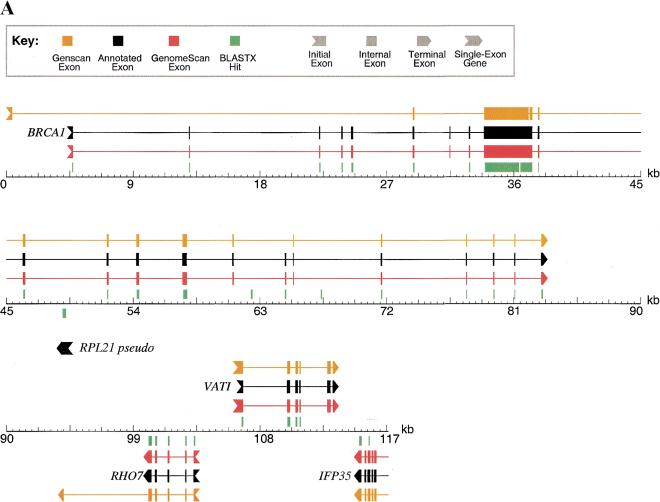
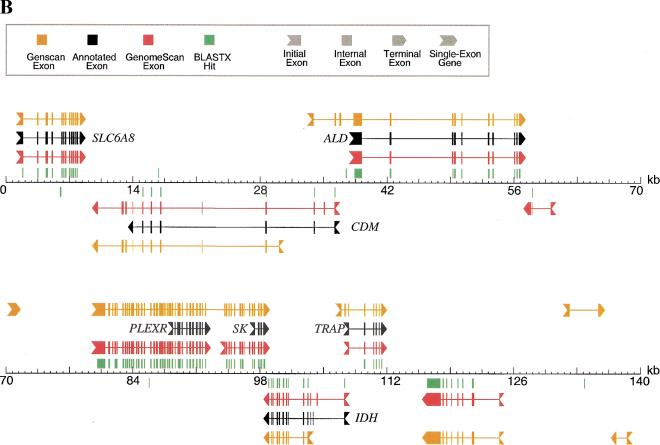
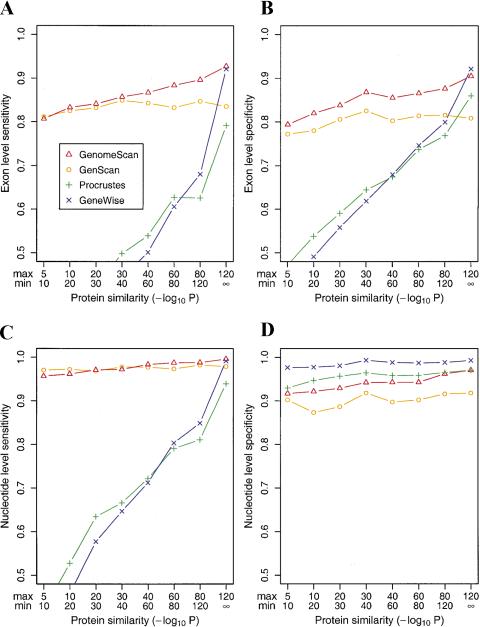
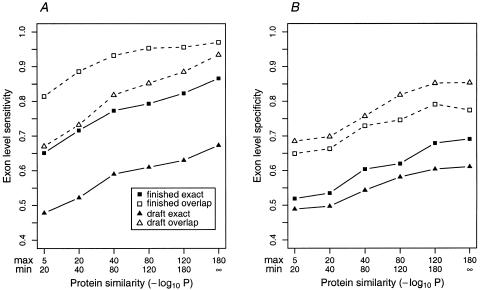
With the human genome sequence approaching completion, a major challenge is to identify the locations and encoded protein sequences of all human genes. To address this problem we have developed a new gene identification algorithm, GenomeScan, which combines exon-intron and splice signal models with similarity to known protein sequences in an integrated model. Extensive testing shows that GenomeScan can accurately identify the exon-intron structures of genes in finished or draft human genome sequence with a low rate of false-positives. Application of GenomeScan to 2.7 billion bases of human genomic DNA identified at least 20,000-25,000 human genes out of an estimated 30,000-40,000 present in the genome. The results show an accurate and efficient automated approach for identifying genes in higher eukaryotic genomes and provide a first-level annotation of the draft human genome.
Figures
References
-
- Brenner V, Nyakatura G, Rosenthal A, Platzer M. Genomic organization of two novel genes on human Xq28: Compact head to head arrangement of IDH gamma and TRAP delta is conserved in rat and mouse. Genomics. 1997;44:8–14. - PubMed
-
- Burge CB. “Identification of genes in human genomic DNA.” Ph.D. thesis. California: Stanford University; 1997.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical