

Human-ovine comparative sequencing of a 250-kb imprinted domain encompassing the callipyge (clpg) locus and identification of six imprinted transcripts: DLK1, DAT, GTL2, PEG11, antiPEG11, and MEG8
- PMID: 11337479
- PMCID: PMC311092
- DOI: 10.1101/gr.172701
Human-ovine comparative sequencing of a 250-kb imprinted domain encompassing the callipyge (clpg) locus and identification of six imprinted transcripts: DLK1, DAT, GTL2, PEG11, antiPEG11, and MEG8
Abstract
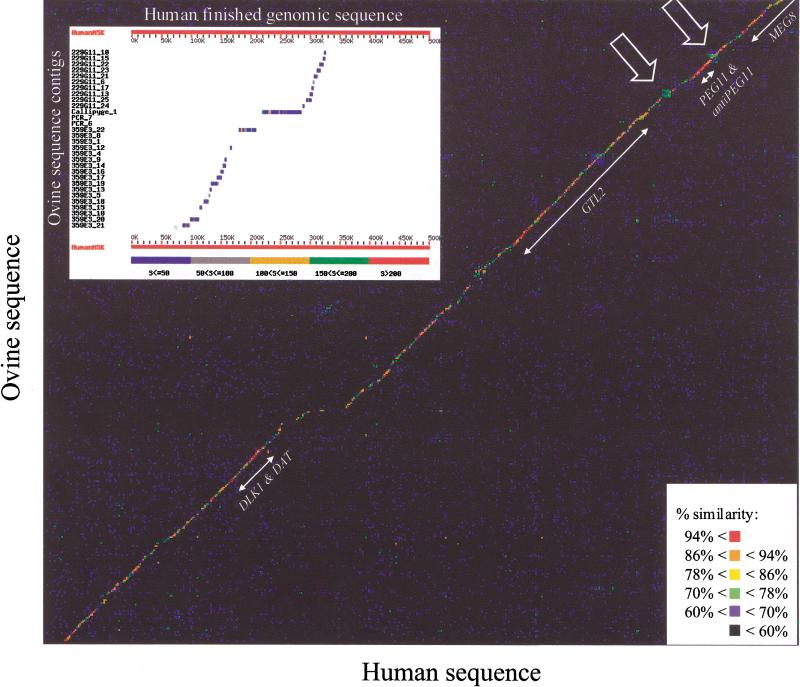
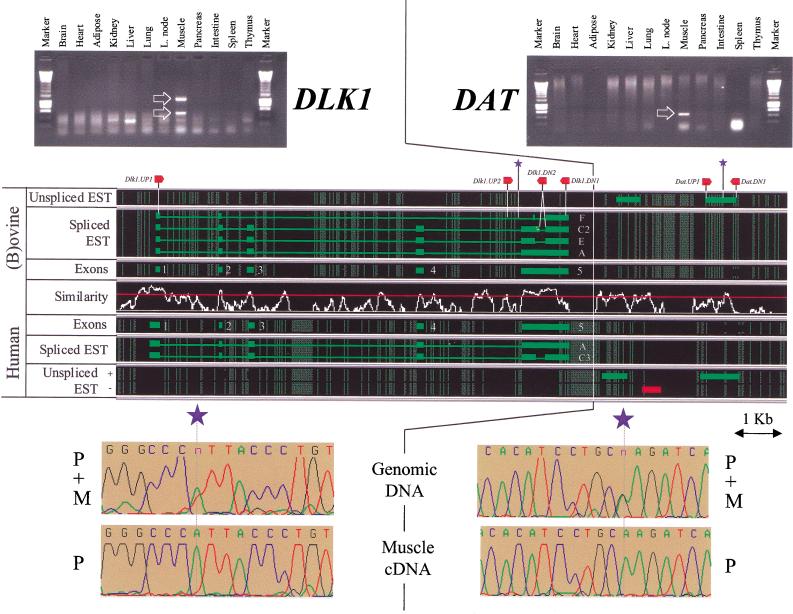
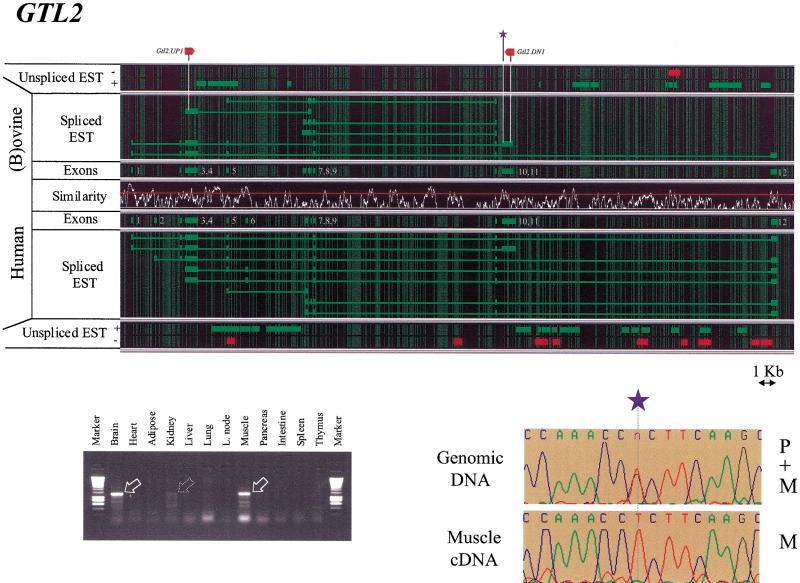
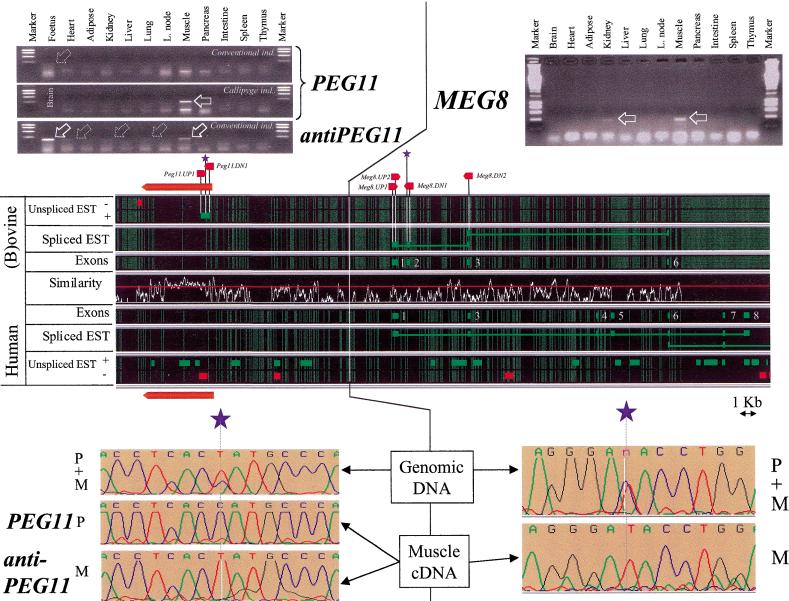
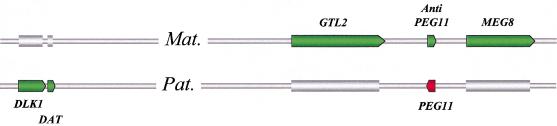
Two ovine BAC clones and a connecting long-range PCR product, jointly spanning approximately 250 kb and representing most of the MULGE5-OY3 marker interval known to contain the clpg locus, were completely sequenced. The resulting genomic sequence was aligned with its human ortholog and extensively annotated. Six transcripts, four of which were novel, were predicted to originate from within the analyzed region and their existence confirmed experimentally: DLK1, DAT, GTL2, PEG11, antiPEG11, and MEG8. RT-PCR experiments performed on a range of tissues sampled from an 8-wk-old animal demonstrated the preferential expression of all six transcripts in skeletal muscle, which suggests that they are under control of common regulatory elements. The six transcripts were also shown to be subject to parental imprinting: DLK1, DAT, and PEG11 were shown to be paternally expressed and GTL2, antiPEG11, and MEG8 to be maternally expressed.
Figures

References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
-
- Berghmans S, Segers K, Shay T, Georges M, Cockett N E, Charlier C. Breakpoint mapping positions the callipyge gene within a 285 kilobase chromosome segment containing the Gtl-2 gene. Mamm Genome. 2001;12:183–185. - PubMed
-
- Bernardi G. The human genome: Organization and evolutionary history. Annu Rev Genet. 1995;29:445–476. - PubMed
-
- Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources