

Basecalling with LifeTrace
- PMID: 11337481
- PMCID: PMC311100
- DOI: 10.1101/gr.177901
Basecalling with LifeTrace
Abstract
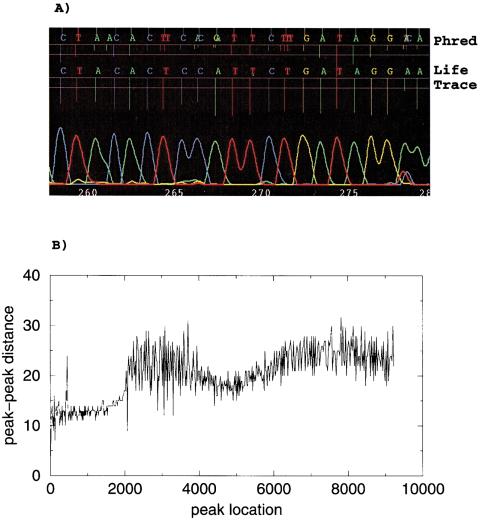
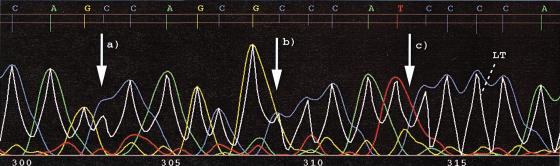
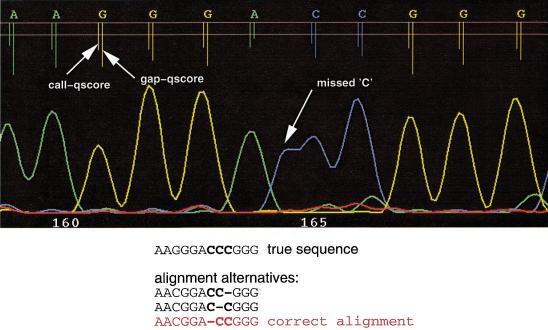
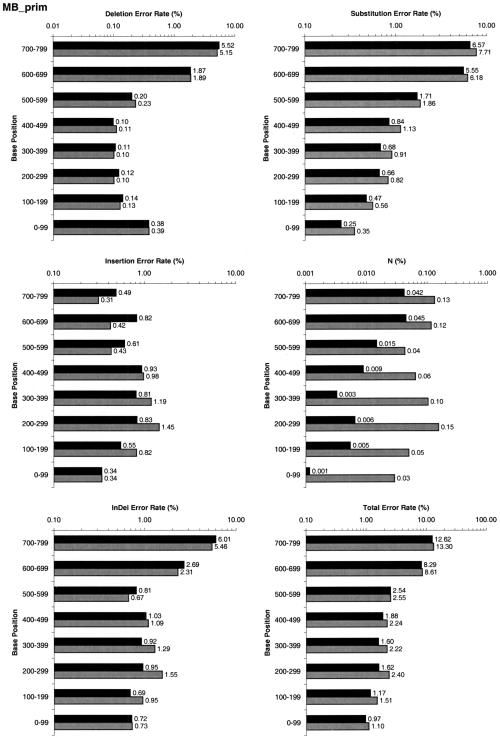
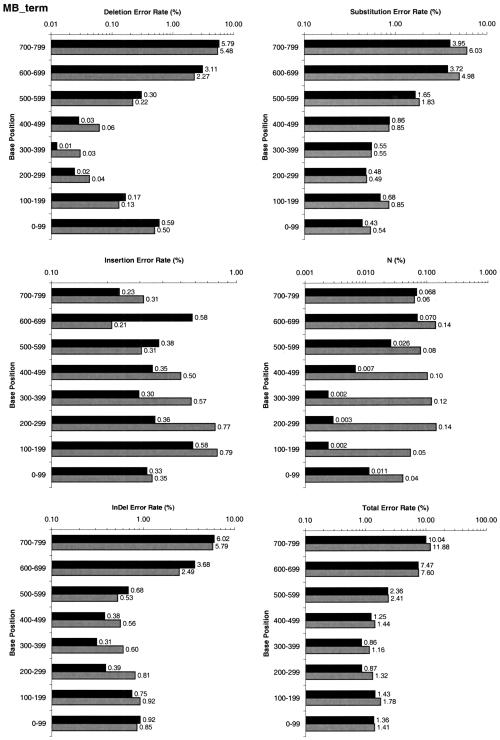
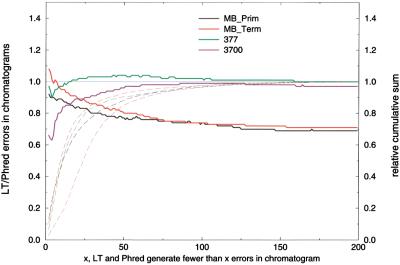
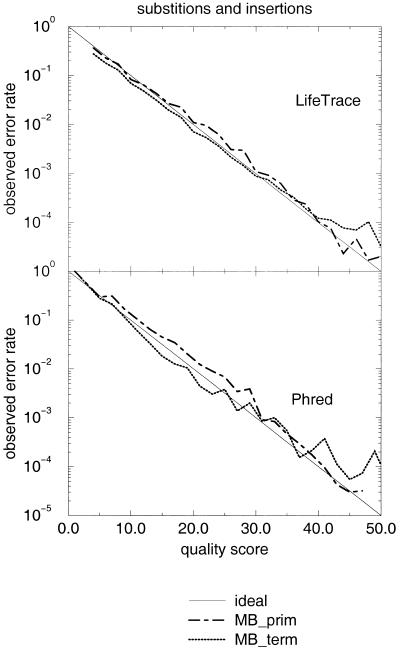
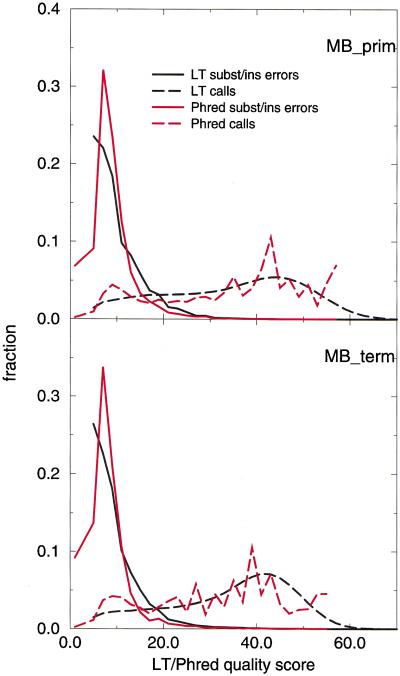
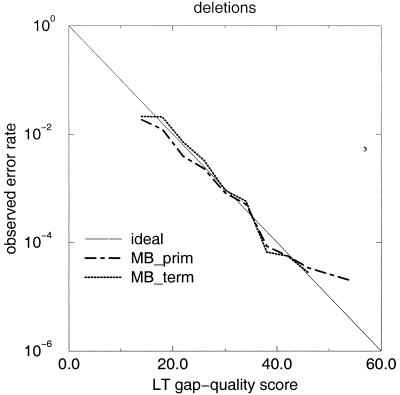
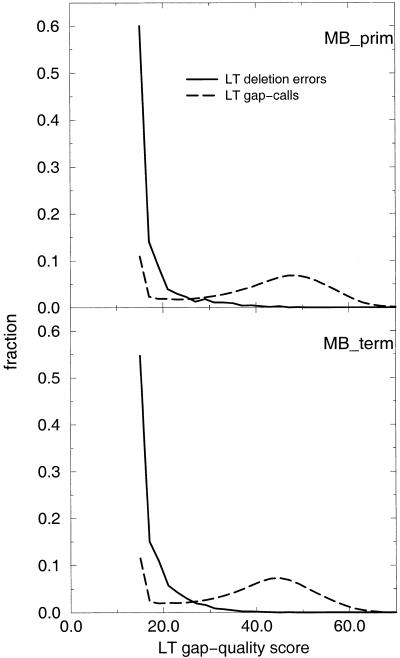
A pivotal step in electrophoresis sequencing is the conversion of the raw, continuous chromatogram data into the actual sequence of discrete nucleotides, a process referred to as basecalling. We describe a novel algorithm for basecalling implemented in the program LifeTrace. Like Phred, currently the most widely used basecalling software program, LifeTrace takes processed trace data as input. It was designed to be tolerant to variable peak spacing by means of an improved peak-detection algorithm that emphasizes local chromatogram information over global properties. LifeTrace is shown to generate high-quality basecalls and reliable quality scores. It proved particularly effective when applied to MegaBACE capillary sequencing machines. In a benchmark test of 8372 dye-primer MegaBACE chromatograms, LifeTrace generated 17% fewer substitution errors, 16% fewer insertion/deletion errors, and 2.4% more aligned bases to the finished sequence than did Phred. For two sets totaling 6624 dye-terminator chromatograms, the performance improvement was 15% fewer substitution errors, 10% fewer insertion/deletion errors, and 2.1% more aligned bases. The processing time required by LifeTrace is comparable to that of Phred. The predicted quality scores were in line with observed quality scores, permitting direct use for quality clipping and in silico single nucleotide polymorphism (SNP) detection. Furthermore, we introduce a new type of quality score associated with every basecall: the gap-quality. It estimates the probability of a deletion error between the current and the following basecall. This additional quality score improves detection of single basepair deletions when used for locating potential basecalling errors during the alignment. We also describe a new protocol for benchmarking that we believe better discerns basecaller performance differences than methods previously published.
Figures
References
-
- Altschul SF, Gish W, Miller W, Myers E, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
-
- Altshuler D, Pollara VJ, Cowles CR, Van Etten WJ, Baldwin J, Linton L, Lander ES. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature. 2000;407:513–516. - PubMed
-
- Berno AJ. A graph theoretic approach to the analysis of DNA sequencing data. Genome Res. 1996;6:90–91. - PubMed
-
- Buetow KH, Edmonson MN, Cassidy AB. Reliable identification of large numbers of candidate SNPs from public EST data. Nat Genet. 1999;21:323–325. - PubMed
-
- Ewing B, Green P. Base-calling of automated sequencer traces using Phred II: Error probabilities. Genome Res. 1998;8:186–194. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous