

Gene structure prediction and alternative splicing analysis using genomically aligned ESTs
- PMID: 11337482
- PMCID: PMC311065
- DOI: 10.1101/gr.155001
Gene structure prediction and alternative splicing analysis using genomically aligned ESTs
Abstract
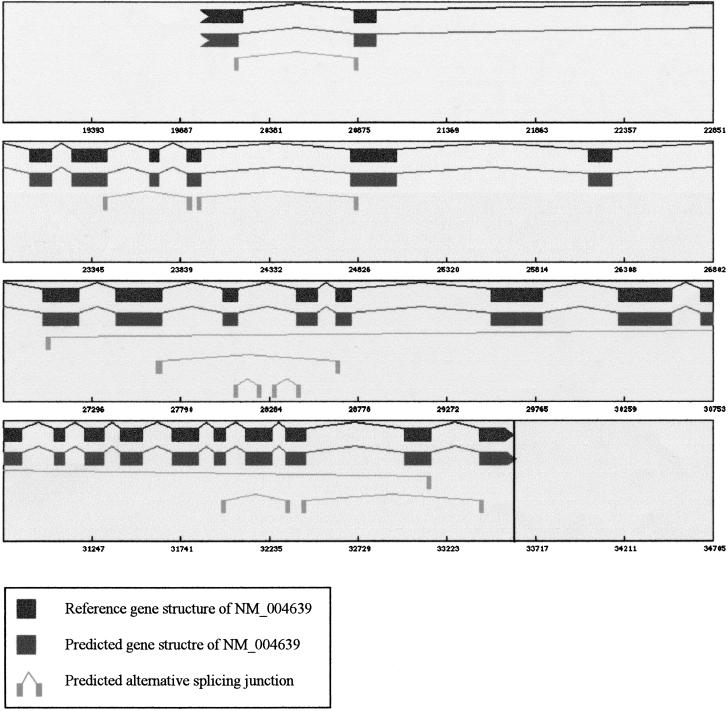
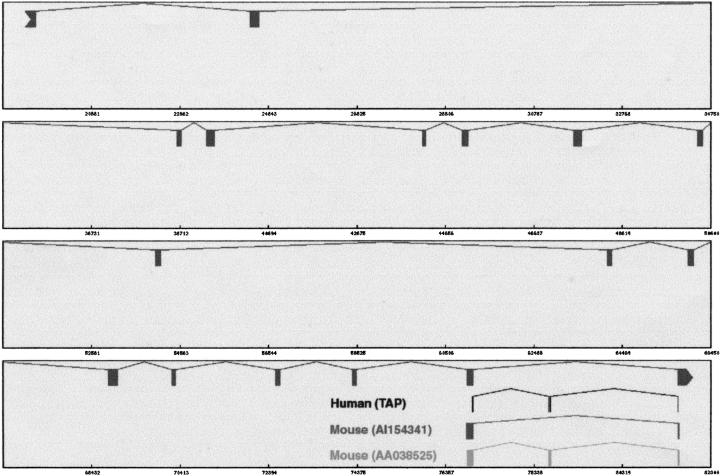
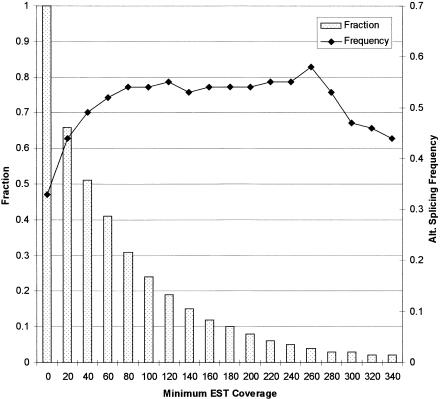
With the availability of a nearly complete sequence of the human genome, aligning expressed sequence tags (EST) to the genomic sequence has become a practical and powerful strategy for gene prediction. Elucidating gene structure is a complex problem requiring the identification of splice junctions, gene boundaries, and alternative splicing variants. We have developed a software tool, Transcript Assembly Program (TAP), to delineate gene structures using genomically aligned EST sequences. TAP assembles the joint gene structure of the entire genomic region from individual splice junction pairs, using a novel algorithm that uses the EST-encoded connectivity and redundancy information to sort out the complex alternative splicing patterns. A method called polyadenylation site scan (PASS) has been developed to detect poly-A sites in the genome. TAP uses these predictions to identify gene boundaries by segmenting the joint gene structure at polyadenylated terminal exons. Reconstructing 1007 known transcripts, TAP scored a sensitivity (Sn) of 60% and a specificity (Sp) of 92% at the exon level. The gene boundary identification process was found to be accurate 78% of the time. also reports alternative splicing patterns in EST alignments. An analysis of alternative splicing in 1124 genic regions suggested that more than half of human genes undergo alternative splicing. Surprisingly, we saw an absolute majority of the detected alternative splicing events affect the coding region. Furthermore, the evolutionary conservation of alternative splicing between human and mouse was analyzed using an EST-based approach. (See http://stl.wustl.edu/~zkan/TAP/)
Figures



References
-
- Bafna V, Huson DH. The conserved exon method for gene finding. Intell Syst Mol Biol. 2000;8:3–12. - PubMed
-
- Bedell JA, Korf I, Gish W. MaskerAid: A performance enhancement to RepeatMasker. Bioinformatics. 2000;16:1040–1041. - PubMed
-
- Bouck J, Yu W, Gibbs R, Worley K. Comparison of gene indexing databases. Trends Genet. 1999;15:159–161. - PubMed
-
- Brett D, Hanke J, Lehmann G, Hasse S, Delbruck S, Krueger S, Reich J, Bork P. EST comparison indicates 38% of human mRNAs contain possible alternative splice forms. FEBS Lett. 2000;47:83–86. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous