

Targeted mutagenesis of Lis1 disrupts cortical development and LIS1 homodimerization
- PMID: 11344260
- PMCID: PMC33485
- DOI: 10.1073/pnas.101122598
Targeted mutagenesis of Lis1 disrupts cortical development and LIS1 homodimerization
Abstract
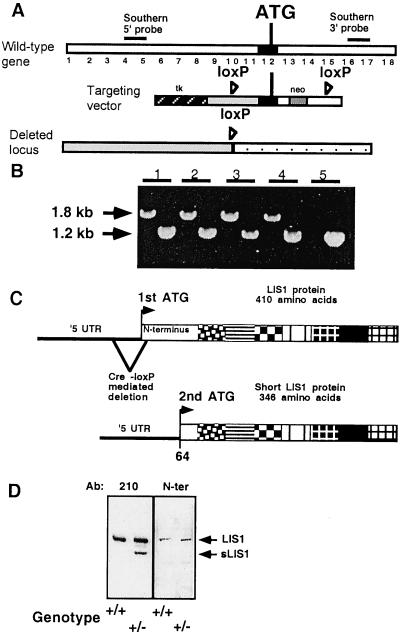
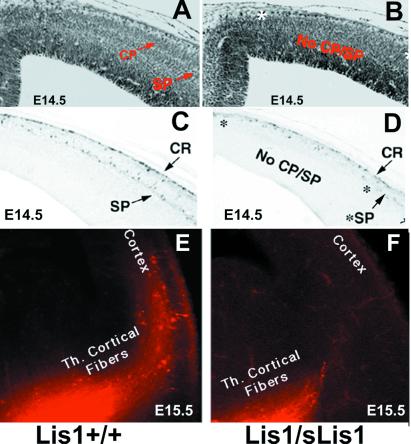
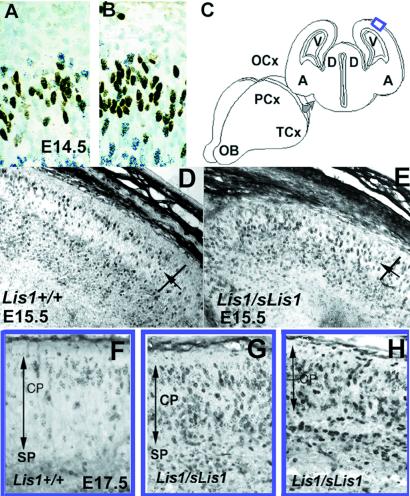
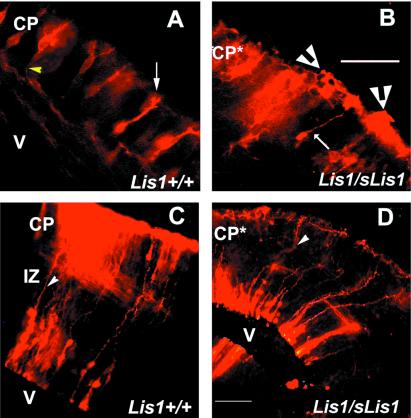
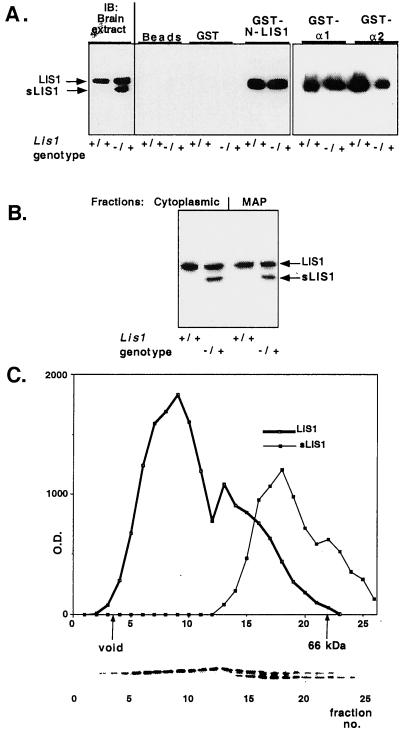
Lissencephaly is a severe brain malformation in humans. To study the function of the gene mutated in lissencephaly (LIS1), we deleted the first coding exon from the mouse Lis1 gene. The deletion resulted in a shorter protein (sLIS1) that initiates from the second methionine, a unique situation because most LIS1 mutations result in a null allele. This mutation mimics a mutation described in one lissencephaly patient with a milder phenotype. Homozygotes are early lethal, although heterozygotes are viable and fertile. Most strikingly, the morphology of cortical neurons and radial glia is aberrant in the developing cortex, and the neurons migrate more slowly. This is the first demonstration, to our knowledge, of a cellular abnormality in the migrating neurons after Lis1 mutation. Moreover, cortical plate splitting and thalomocortical innervation are also abnormal. Biochemically, the mutant protein is not capable of dimerization, and enzymatic activity is elevated in the embryos, thus a demonstration of the in vivo role of LIS1 as a subunit of PAF-AH. This mutation allows us to determine a hierarchy of functions that are sensitive to LIS1 dosage, thus promoting our understanding of the role of LIS1 in the developing cortex.
Figures
References
-
- Reiner O, Carrozzo R, Shen Y, Whenert M, Faustinella F, Dobyns W B, Caskey C T, Ledbetter D H. Nature (London) 1993;364:717–721. - PubMed
-
- Reiner O. Mol Neurobiol. 2000;20:143–156. - PubMed
-
- des Portes V, Pinard J M, Billuart P, Vinet M C, Koulakoff A, Carrie A, Gelot A, Dupuis E, Motte J, Berwald-Netter Y, et al. Cell. 1998;92:51–61. - PubMed
-
- Gleeson J G, Allen K M, Fox J W, Lamperti E D, Berkovic S, Scheffer I, Cooper E C, Dobyns W B, Minnerath S R, Ross M E, Walsh C A. Cell. 1998;92:63–72. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
