

Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia
- PMID: 11344269
- PMCID: PMC33497
- DOI: 10.1073/pnas.101545198
Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia
Abstract
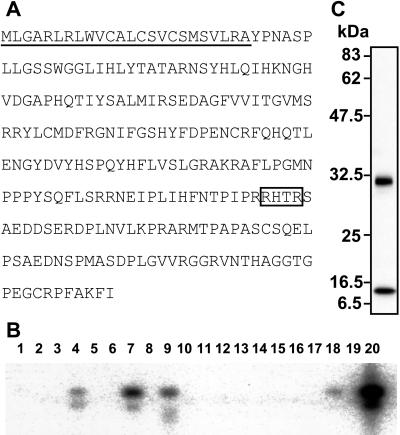
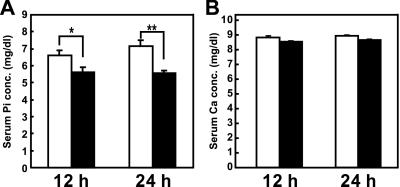
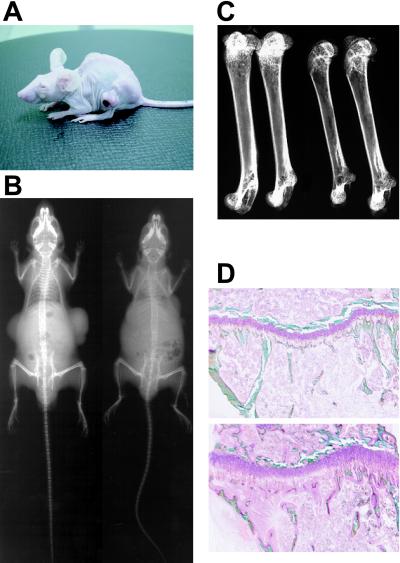
Tumor-induced osteomalacia (TIO) is one of the paraneoplastic diseases characterized by hypophosphatemia caused by renal phosphate wasting. Because removal of responsible tumors normalizes phosphate metabolism, an unidentified humoral phosphaturic factor is believed to be responsible for this syndrome. To identify the causative factor of TIO, we obtained cDNA clones that were abundantly expressed only in a tumor causing TIO and constructed tumor-specific cDNA contigs. Based on the sequence of one major contig, we cloned 2,270-bp cDNA, which turned out to encode fibroblast growth factor 23 (FGF23). Administration of recombinant FGF23 decreased serum phosphate in mice within 12 h. When Chinese hamster ovary cells stably expressing FGF23 were s.c. implanted into nude mice, hypophosphatemia with increased renal phosphate clearance was observed. In addition, a high level of serum alkaline phosphatase, low 1,25-dihydroxyvitamin D, deformity of bone, and impairment of body weight gain became evident. Histological examination showed marked increase of osteoid and widening of growth plate. Thus, continuous production of FGF23 reproduced clinical, biochemical, and histological features of TIO in vivo. Analyses for recombinant FGF23 products produced by Chinese hamster ovary cells indicated proteolytic cleavage of FGF23 at the RXXR motif. Recent genetic study indicates that missense mutations in this RXXR motif of FGF23 are responsible for autosomal dominant hypophosphatemic rickets, another hypophosphatemic disease with similar features to TIO. We conclude that overproduction of FGF23 causes TIO, whereas mutations in the FGF23 gene result in autosomal dominant hypophosphatemic rickets possibly by preventing proteolytic cleavage and enhancing biological activity of FGF23.
Figures


Comment in
-
FGF23, hypophosphatemia, and rickets: has phosphatonin been found?Proc Natl Acad Sci U S A. 2001 May 22;98(11):5945-6. doi: 10.1073/pnas.111154898. Proc Natl Acad Sci U S A. 2001. PMID: 11371627 Free PMC article. No abstract available.
References
-
- Drezner M K. In: Primer on Metabolic Bone Diseases and Disorders of Mineral Metabolism. Favus M J, editor. Philadelphia: Lippincott; 1999. pp. 331–337.
-
- Kumar R. Bone. 2000;27:333–338. - PubMed
-
- Cai Q, Hodgson S F, Kao P C, Lennon V A, Klee G G, Zinsmiester A R, Kumar R. N Engl J Med. 1994;330:1645–1649. - PubMed
-
- Wilkins G E, Granleese S, Hegele R G, Holden J, Anderson D W, Bondy G P. J Clin Endocrinol Metab. 1995;80:1628–1634. - PubMed
-
- Nelson A E, Namkung H J, Patava J, Wilkinson M R, Chang A C, Reddel R R, Robinson B G, Mason R S. Mol Cell Endocrinol. 1996;124:17–23. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
