

Identification of intrinsic order and disorder in the DNA repair protein XPA
- PMID: 11344324
- PMCID: PMC2374143
- DOI: 10.1110/ps.29401
Identification of intrinsic order and disorder in the DNA repair protein XPA
Abstract
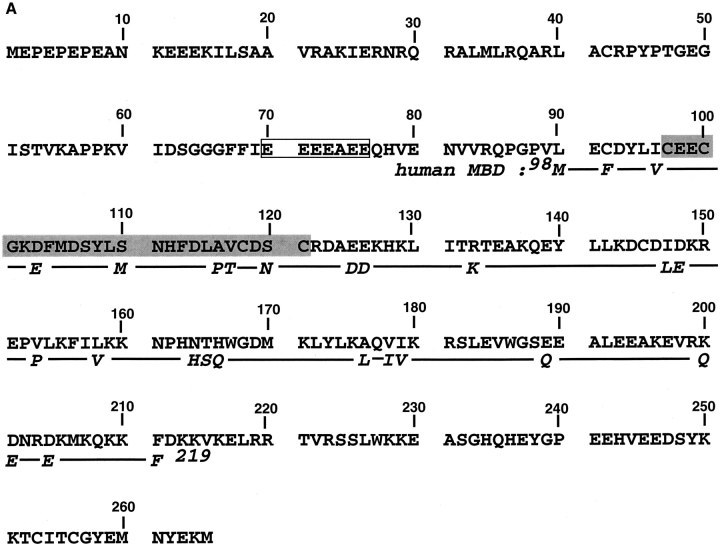
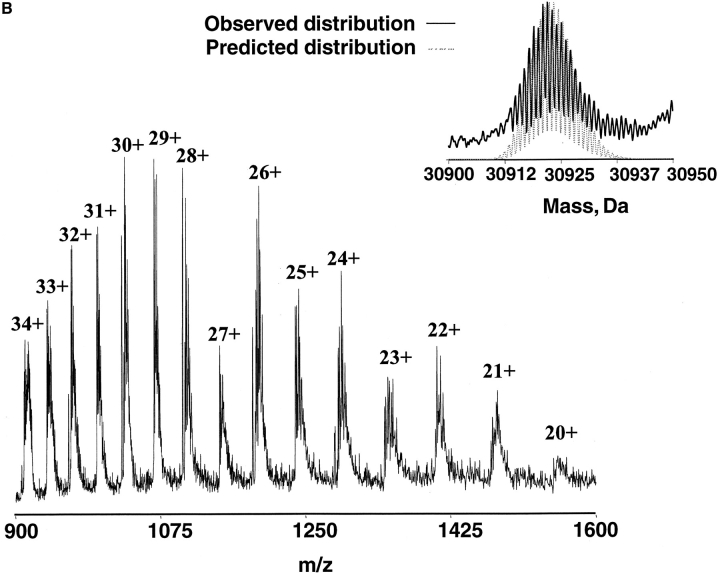
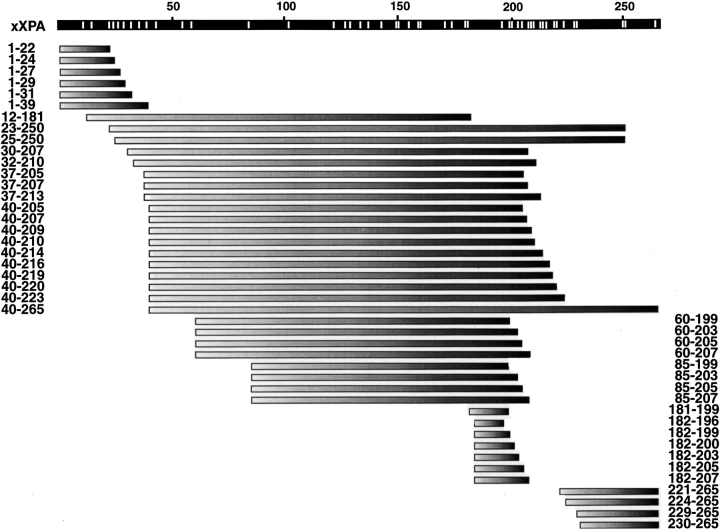
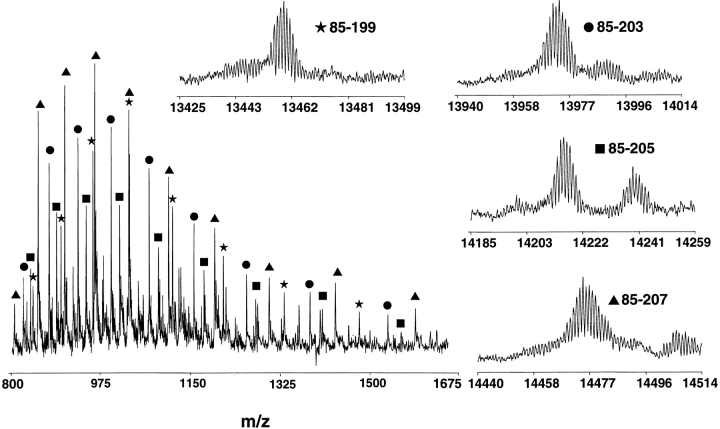
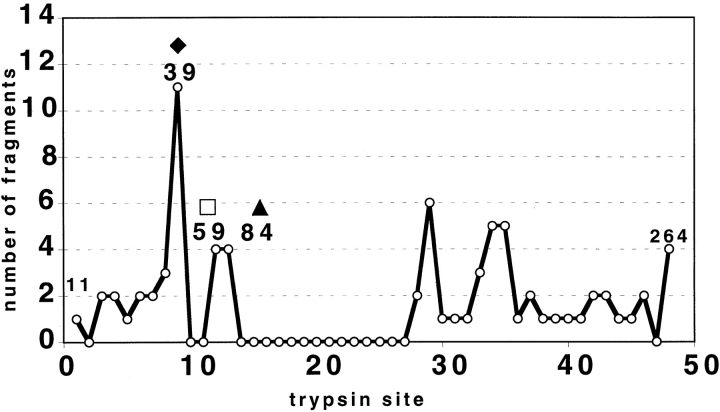
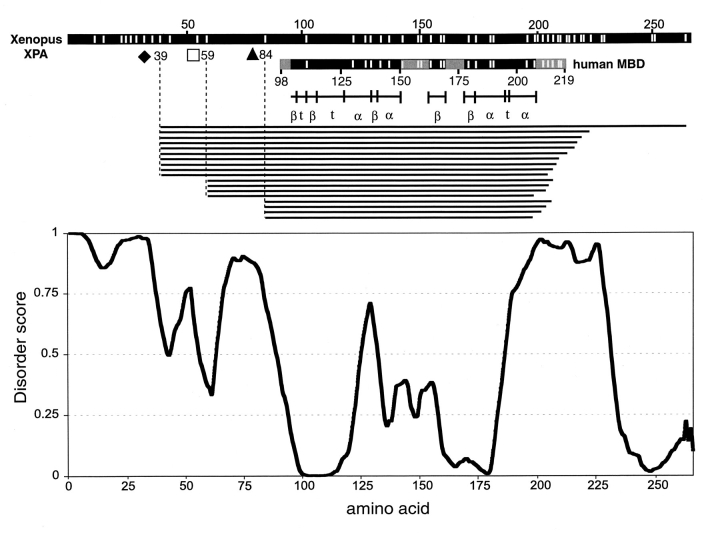
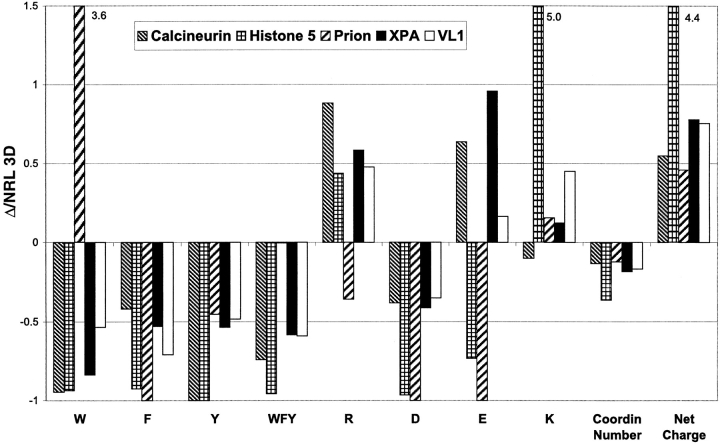
The DNA-repair protein XPA is required to recognize a wide variety of bulky lesions during nucleotide excision repair. Independent NMR solution structures of a human XPA fragment comprising approximately 40% of the full-length protein, the minimal DNA-binding domain, revealed that one-third of this molecule was disordered. To better characterize structural features of full-length XPA, we performed time-resolved trypsin proteolysis on active recombinant Xenopus XPA (xXPA). The resulting proteolytic fragments were analyzed by electrospray ionization interface coupled to a Fourier transform ion cyclotron resonance mass spectrometry and SDS-PAGE. The molecular weight of the full-length xXPA determined by mass spectrometry (30922.02 daltons) was consistent with that calculated from the sequence (30922.45 daltons). Moreover, the mass spectrometric data allowed the assignment of multiple xXPA fragments not resolvable by SDS-PAGE. The neural network program Predictor of Natural Disordered Regions (PONDR) applied to xXPA predicted extended disordered N- and C-terminal regions with an ordered internal core. This prediction agreed with our partial proteolysis results, thereby indicating that disorder in XPA shares sequence features with other well-characterized intrinsically unstructured proteins. Trypsin cleavages at 30 of the possible 48 sites were detected and no cleavage was observed in an internal region (Q85-I179) despite 14 possible cut sites. For the full-length xXPA, there was strong agreement among PONDR, partial proteolysis data, and the NMR structure for the corresponding XPA fragment.
Figures


Similar articles
-
Aberrant mobility phenomena of the DNA repair protein XPA.Protein Sci. 2001 Jul;10(7):1353-62. doi: 10.1110/ps.ps.40101. Protein Sci. 2001. PMID: 11420437 Free PMC article.
-
Extended X-ray absorption fine structure evidence for a single metal binding domain in Xenopus laevis nucleotide excision repair protein XPA.Biochem Biophys Res Commun. 1999 Jan 8;254(1):109-13. doi: 10.1006/bbrc.1998.9890. Biochem Biophys Res Commun. 1999. PMID: 9920741
-
Human nucleotide excision repair protein XPA: 1H NMR and CD solution studies of a synthetic peptide fragment corresponding to the zinc-binding domain (101-141).J Biomol Struct Dyn. 1997 Jun;14(6):677-90. doi: 10.1080/07391102.1997.10508171. J Biomol Struct Dyn. 1997. PMID: 9195337
-
Equilibrium and stop-flow kinetic studies of fluorescently labeled DNA substrates with DNA repair proteins XPA and replication protein A.Biochemistry. 2002 Jan 8;41(1):131-43. doi: 10.1021/bi011041q. Biochemistry. 2002. PMID: 11772010
-
Electrospray ionization-mass spectrometry study of the interaction of cisplatin-adducted oligonucleotides with human XPA minimal binding domain protein.Anal Biochem. 1999 Jul 15;272(1):26-33. doi: 10.1006/abio.1999.4143. Anal Biochem. 1999. PMID: 10405289
Cited by
-
Wrecked regulation of intrinsically disordered proteins in diseases: pathogenicity of deregulated regulators.Front Mol Biosci. 2014 Jul 25;1:6. doi: 10.3389/fmolb.2014.00006. eCollection 2014. Front Mol Biosci. 2014. PMID: 25988147 Free PMC article. Review.
-
Intrinsic disorder and functional proteomics.Biophys J. 2007 Mar 1;92(5):1439-56. doi: 10.1529/biophysj.106.094045. Epub 2006 Dec 8. Biophys J. 2007. PMID: 17158572 Free PMC article. Review.
-
The ERC1 scaffold protein implicated in cell motility drives the assembly of a liquid phase.Sci Rep. 2019 Sep 19;9(1):13530. doi: 10.1038/s41598-019-49630-y. Sci Rep. 2019. PMID: 31537859 Free PMC article.
-
Cross-species investigation into the requirement of XPA for nucleotide excision repair.Nucleic Acids Res. 2024 Jan 25;52(2):677-689. doi: 10.1093/nar/gkad1104. Nucleic Acids Res. 2024. PMID: 37994737 Free PMC article.
-
Paradoxes and wonders of intrinsic disorder: Stability of instability.Intrinsically Disord Proteins. 2017 Oct 16;5(1):e1327757. doi: 10.1080/21690707.2017.1327757. eCollection 2017. Intrinsically Disord Proteins. 2017. PMID: 30250771 Free PMC article.
References
-
- Ackerman, E.J. and Iakoucheva, L.M. 2000. Nucleotide excision repair in oocyte nuclear extracts from Xenopus laevis. Methods: Companion Meth. Enzymol. 22 : 188–193. - PubMed
-
- Araujo, S.J. and Wood, R.D. 1999. Protein complexes in nucleotide excision repair. Mutation Res. 435 23–33. - PubMed
-
- Aviles, F.J., Chapman, G.E., Kneale, G.G., Crane-Robinson, C., and Bradbury, E.M. 1978. The conformation of histone H5. Isolation and characterisation of the globular segment. Eur. J. Biochem. 88 363–371. - PubMed
-
- Bode, W., Schwager, P., and Huber, R. 1978. The transition of bovine trypsinogen to a trypsin-like state upon strong ligand binding. The refined crystal structures of the bovine trypsinogen-pancreatic trypsin inhibitor complex and of its ternary complex with Ile-Val at 1.9 A resolution. J. Mol. Biol. 118 99–112. - PubMed
-
- Bothner, B., Dong, X.F., Bibbs, L., Johnson, J.E., and Siuzdak, G. 1998. Evidence of viral capsid dynamics using limited proteolysis and mass spectrometry. J. Biol. Chem. 273 673–676. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
