

Discovering common stem-loop motifs in unaligned RNA sequences
- PMID: 11353083
- PMCID: PMC55461
- DOI: 10.1093/nar/29.10.2135
Discovering common stem-loop motifs in unaligned RNA sequences
Abstract
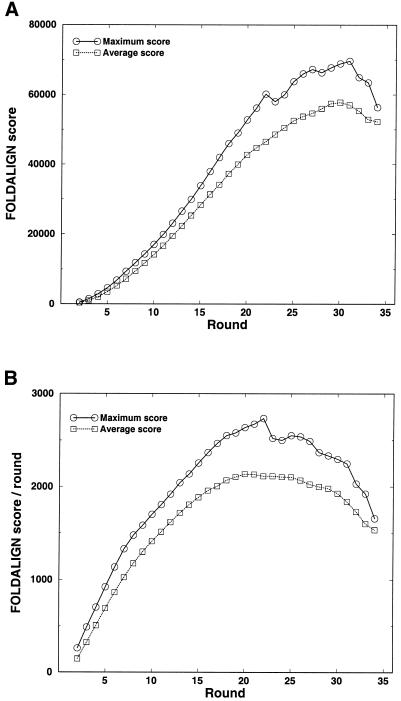
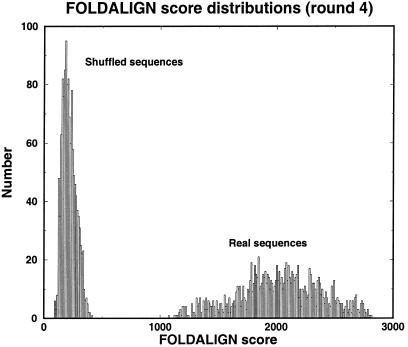
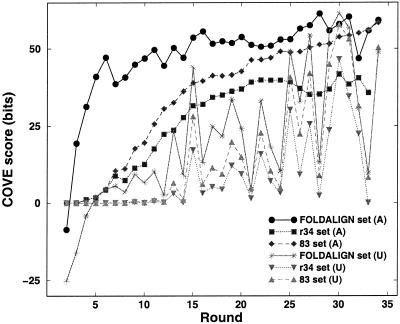
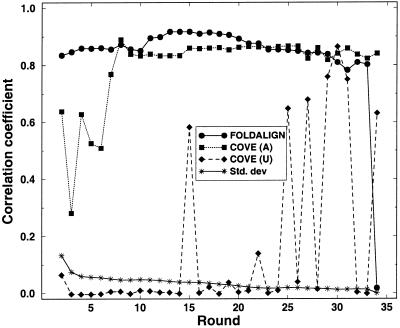
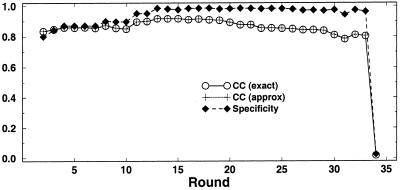
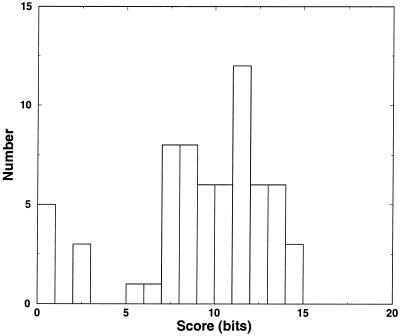
Post-transcriptional regulation of gene expression is often accomplished by proteins binding to specific sequence motifs in mRNA molecules, to affect their translation or stability. The motifs are often composed of a combination of sequence and structural constraints such that the overall structure is preserved even though much of the primary sequence is variable. While several methods exist to discover transcriptional regulatory sites in the DNA sequences of coregulated genes, the RNA motif discovery problem is much more difficult because of covariation in the positions. We describe the combined use of two approaches for RNA structure prediction, FOLDALIGN and COVE, that together can discover and model stem-loop RNA motifs in unaligned sequences, such as UTRs from post-transcriptionally coregulated genes. We evaluate the method on two datasets, one a section of rRNA genes with randomly truncated ends so that a global alignment is not possible, and the other a hyper-variable collection of IRE-like elements that were inserted into randomized UTR sequences. In both cases the combined method identified the motifs correctly, and in the rRNA example we show that it is capable of determining the structure, which includes bulge and internal loops as well as a variable length hairpin loop. Those automated results are quantitatively evaluated and found to agree closely with structures contained in curated databases, with correlation coefficients up to 0.9. A basic server, Stem-Loop Align SearcH (SLASH), which will perform stem-loop searches in unaligned RNA sequences, is available at http://www.bioinf.au.dk/slash/.
Figures


References
-
- Pabo C.O. and Nekludova,L. (2000) Geometric analysis and comparison of protein–DNA interfaces: why is there no simple code for recognition? J. Mol. Biol., 301, 597–624. - PubMed
-
- Gray N.K. and Hentze,M.W. (1994) Regulation of protein synthesis by mRNA structure. Mol. Biol. Rep., 19, 195–200. - PubMed
-
- Klaff P., Riesner,D. and Steger,G. (1996) RNA structure and the regulation of gene expression. Plant Mol. Biol., 32, 89–106. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
