

Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1
- PMID: 11359924
- PMCID: PMC34586
- DOI: 10.1091/mbc.12.5.1315
Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1
Abstract
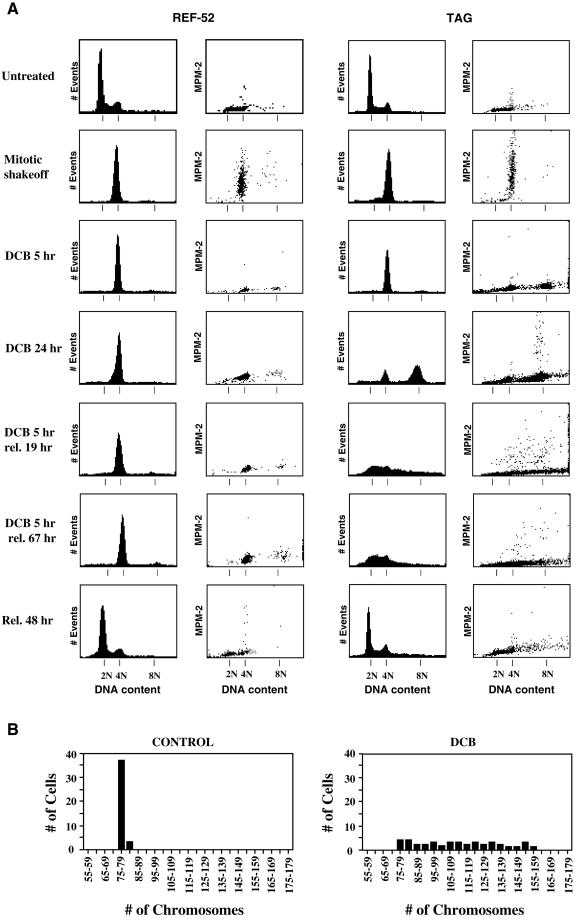
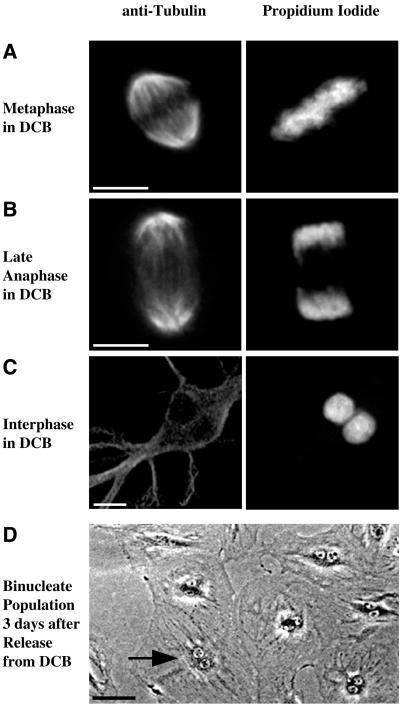
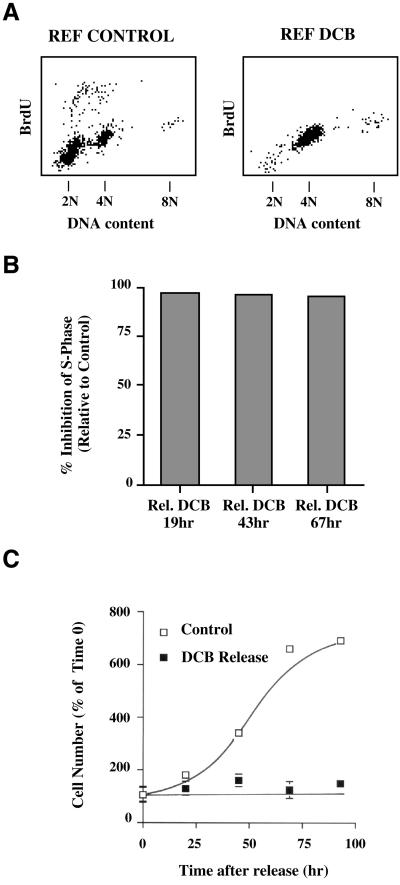
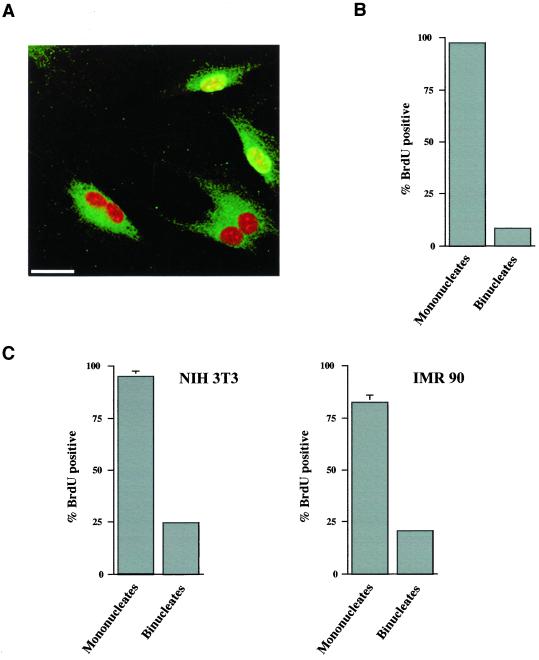
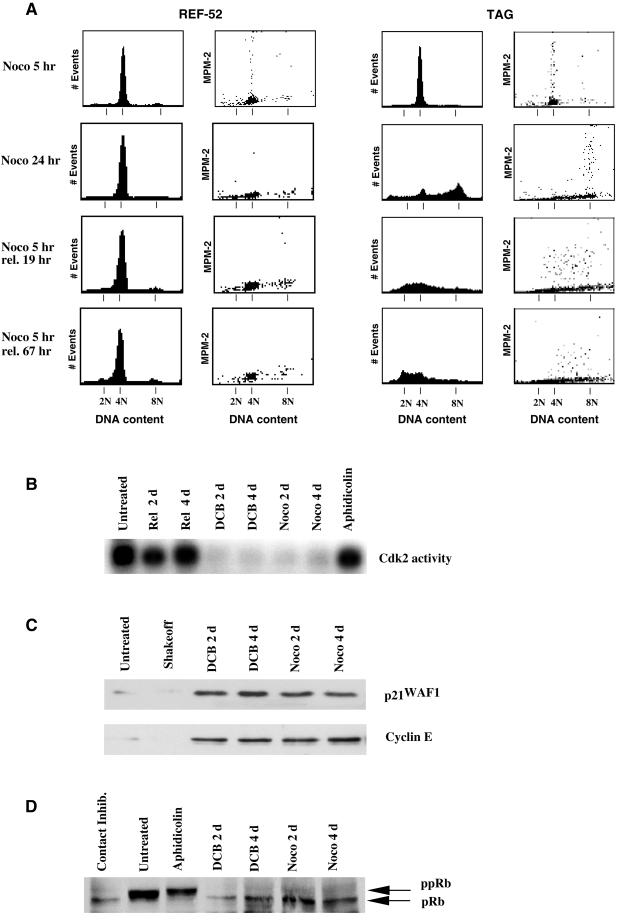
A "spindle assembly" checkpoint has been described that arrests cells in G1 following inappropriate exit from mitosis in the presence of microtubule inhibitors. We have here addressed the question of whether the resulting tetraploid state itself, rather than failure of spindle function or induction of spindle damage, acts as a checkpoint to arrest cells in G1. Dihydrocytochalasin B induces cleavage failure in cells where spindle function and chromatid segregation are both normal. Notably, we show here that nontransformed REF-52 cells arrest indefinitely in tetraploid G1 following cleavage failure. The spindle assembly checkpoint and the tetraploidization checkpoint that we describe here are likely to be equivalent. Both involve arrest in G1 with inactive cdk2 kinase, hypophosphorylated retinoblastoma protein, and elevated levels of p21(WAF1) and cyclin E. Furthermore, both require p53. We show that failure to arrest in G1 following tetraploidization rapidly results in aneuploidy. Similar tetraploid G1 arrest results have been obtained with mouse NIH3T3 and human IMR-90 cells. Thus, we propose that a general checkpoint control acts in G1 to recognize tetraploid cells and induce their arrest and thereby prevents the propagation of errors of late mitosis and the generation of aneuploidy. As such, the tetraploidy checkpoint may be a critical activity of p53 in its role of ensuring genomic integrity.
Figures
References
-
- Andreassen PR, Martineau SN, Margolis RL. Chemical induction of mitotic checkpoint override in mammalian cells results in aneuploidy following a transient tetraploid state. Mutat Res. 1996;372:181–194. - PubMed
-
- Aubin JE, Osborn M, Weber K. Inhibition of cytokinesis and altered contractile ring morphology induced by cytochalasins in synchronized PtK2 cells. Exp Cell Res. 1981;136:63–79. - PubMed
-
- Baker SJ, Markowitz S, Fearon ER, Willson JK, Vogelstein B. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science. 1990;249:912–915. - PubMed
-
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
