

Segmental duplications: organization and impact within the current human genome project assembly
- PMID: 11381028
- PMCID: PMC311093
- DOI: 10.1101/gr.gr-1871r
Segmental duplications: organization and impact within the current human genome project assembly
Abstract
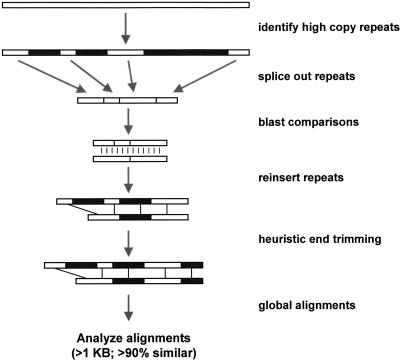
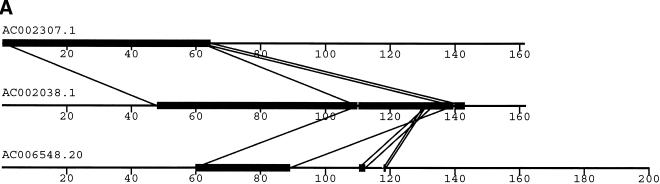
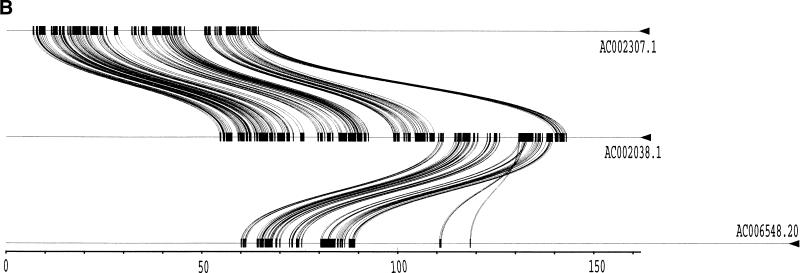
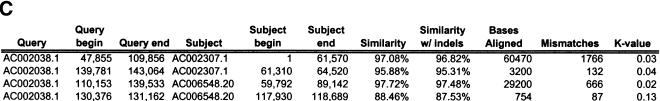
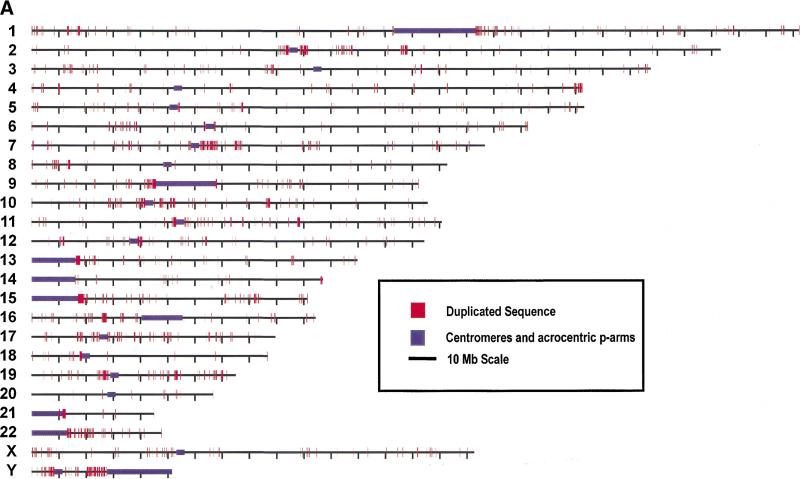
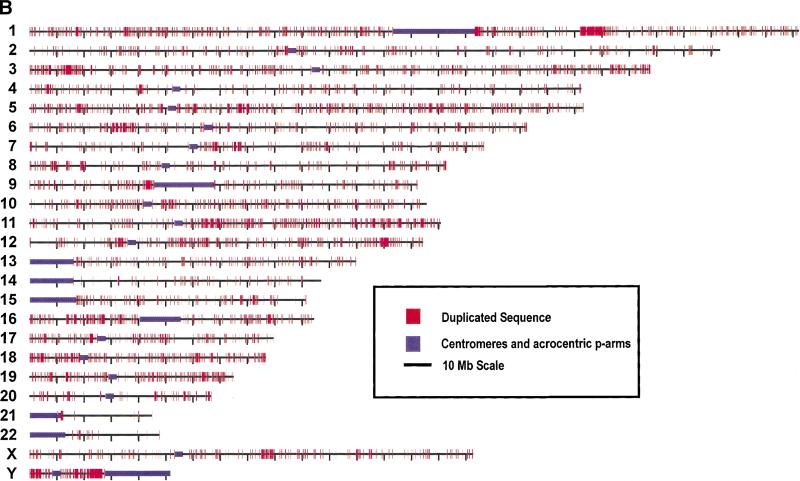
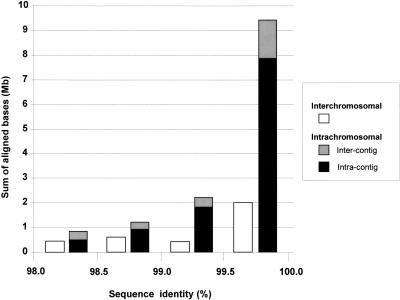
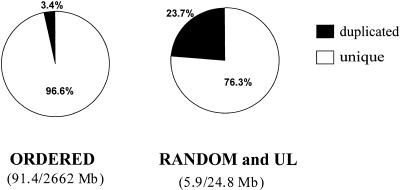
Segmental duplications play fundamental roles in both genomic disease and gene evolution. To understand their organization within the human genome, we have developed the computational tools and methods necessary to detect identity between long stretches of genomic sequence despite the presence of high copy repeats and large insertion-deletions. Here we present our analysis of the most recent genome assembly (January 2001) in which we focus on the global organization of these segments and the role they play in the whole-genome assembly process. Initially, we considered only large recent duplication events that fell well-below levels of draft sequencing error (alignments 90%-98% similar and > or =1 kb in length). Duplications (90%-98%; > or =1 kb) comprise 3.6% of all human sequence. These duplications show clustering and up to 10-fold enrichment within pericentromeric and subtelomeric regions. In terms of assembly, duplicated sequences were found to be over-represented in unordered and unassigned contigs indicating that duplicated sequences are difficult to assign to their proper position. To assess coverage of these regions within the genome, we selected BACs containing interchromosomal duplications and characterized their duplication pattern by FISH. Only 47% (106/224) of chromosomes positive by FISH had a corresponding chromosomal position by comparison. We present data that indicate that this is attributable to misassembly, misassignment, and/or decreased sequencing coverage within duplicated regions. Surprisingly, if we consider putative duplications >98% identity, we identify 10.6% (286 Mb) of the current assembly as paralogous. The majority of these alignments, we believe, represent unmerged overlaps within unique regions. Taken together the above data indicate that segmental duplications represent a significant impediment to accurate human genome assembly, requiring the development of specialized techniques to finish these exceptional regions of the genome. The identification and characterization of these highly duplicated regions represents an important step in the complete sequencing of a human reference genome.
Figures

References
-
- Amann J, Valentine M, Kidd VJ, Lahti JM. Localization of chi1-related helicase genes to human chromosome regions 12p11 and 12p13: Similarity between parts of these genes and conserved human telomeric-associated DNA. Genomics. 1996;32:260–265. - PubMed
-
- Amos-Landgraf JM, Ji Y, Gottlieb W, Depinet T, Wandstrat AE, Cassidy SB, Driscoll DJ, Rogan PK, Schwartz S, Nicholls RD. Chromosome breakage in the Prader-Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. Am J Hum Genet. 1999;65:370–386. - PMC - PubMed
-
- Bentley DR, Deloukas P, Dunham A, French L, Gregory SG, Humphray SJ, Mungall AJ, Ross MT, Carter NP, Dunham I, et al. The physical maps for sequencing human chromosomes 1, 6, 9, 10, 13, 20, and X. Nature. 2001;409:942–943. - PubMed
-
- Brenner S, Elgar G, Sandford R, Macrae A, Venkatesh B, Aparicio S. Characterization of the pufferfish (Fugu) genome as a compact model vertebrate genome. Nature. 1993;366:265–268. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous