

Comprehensive genome sequence analysis of a breast cancer amplicon
- PMID: 11381030
- PMCID: PMC311107
- DOI: 10.1101/gr.gr1743r
Comprehensive genome sequence analysis of a breast cancer amplicon
Abstract
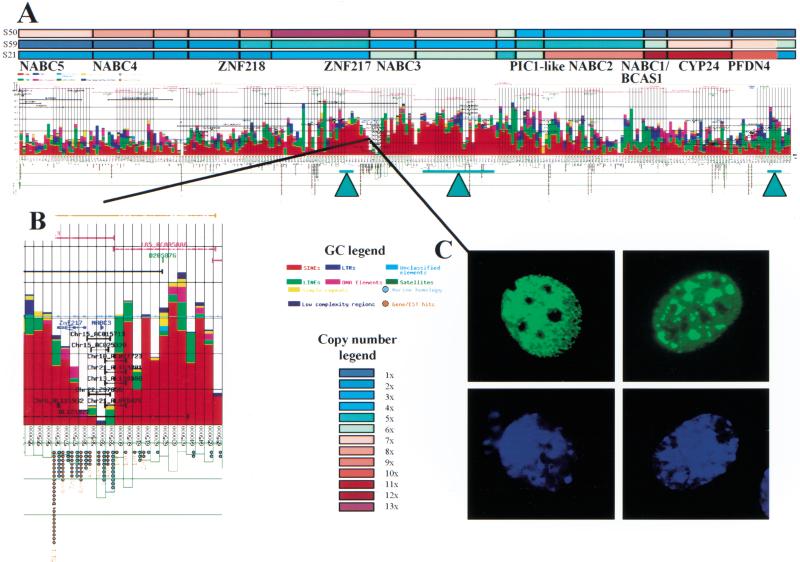
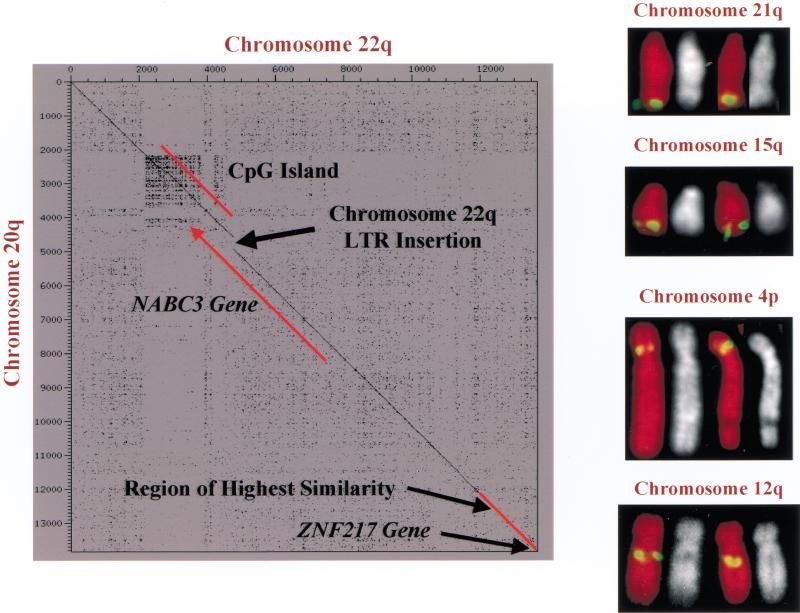
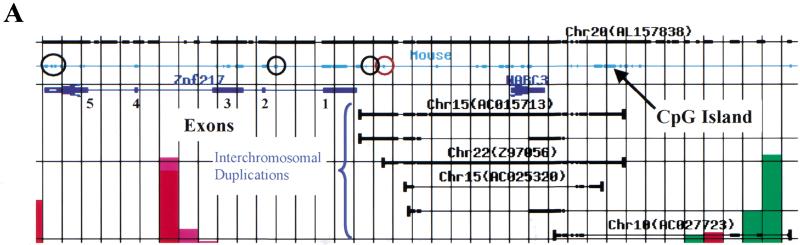
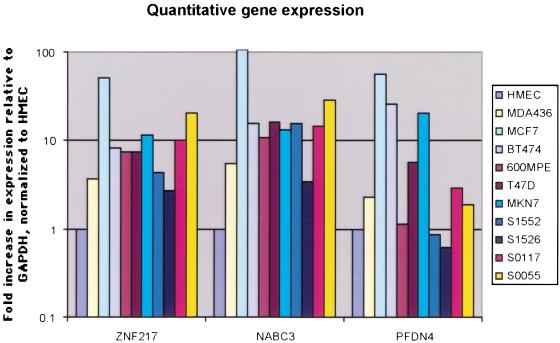
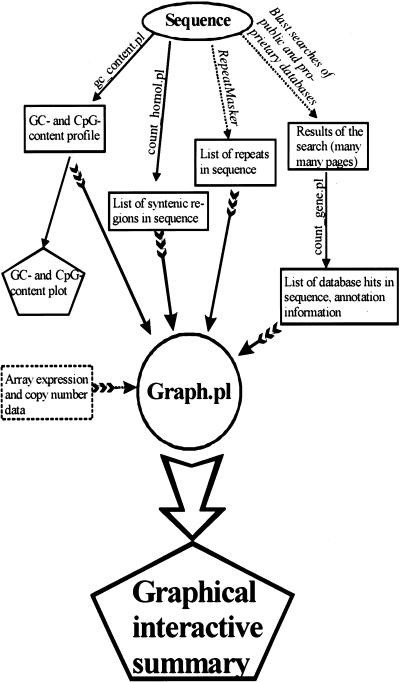
Gene amplification occurs in most solid tumors and is associated with poor prognosis. Amplification of 20q13.2 is common to several tumor types including breast cancer. The 1 Mb of sequence spanning the 20q13.2 breast cancer amplicon is one of the most exhaustively studied segments of the human genome. These studies have included amplicon mapping by comparative genomic hybridization (CGH), fluorescent in-situ hybridization (FISH), array-CGH, quantitative microsatellite analysis (QUMA), and functional genomic studies. Together these studies revealed a complex amplicon structure suggesting the presence of at least two driver genes in some tumors. One of these, ZNF217, is capable of immortalizing human mammary epithelial cells (HMEC) when overexpressed. In addition, we now report the sequencing of this region in human and mouse, and on quantitative expression studies in tumors. Amplicon localization now is straightforward and the availability of human and mouse genomic sequence facilitates their functional analysis. However, comprehensive annotation of megabase-scale regions requires integration of vast amounts of information. We present a system for integrative analysis and demonstrate its utility on 1.2 Mb of sequence spanning the 20q13.2 breast cancer amplicon and 865 kb of syntenic murine sequence. We integrate tumor genome copy number measurements with exhaustive genome landscape mapping, showing that amplicon boundaries are associated with maxima in repetitive element density and a region of evolutionary instability. This integration of comprehensive sequence annotation, quantitative expression analysis, and tumor amplicon boundaries provide evidence for an additional driver gene prefoldin 4 (PFDN4), coregulated genes, conserved noncoding regions, and associate repetitive elements with regions of genomic instability at this locus.
Figures

References
-
- Albertson DG, Ylstra B, Segraves R, Collins C, Dairkee SH, Kowbel D, Kuo WL, Gray JW, Pinkel D. Quantitative mapping of amplicon structure by array CGH identifies CYP24 as a candidate oncogene. Nat Genet. 2000;25:144–146. - PubMed
-
- Beckman MJ, DeLuca HF. Assay of 25-hydroxyvitamin D 1 alpha-hydroxylase and 24-hydroxylase. Methods Enzymol. 1997;282:200–213. - PubMed
-
- Brosius J. RNAs from all categories generate retrosequences that may be exapted as novel genes or regulatory elements. Gene. 1999;238:115–134. - PubMed
-
- Christian SL, Fantes JA, Mewborn SK, Huang B, Ledbetter DH. Large genomic duplicons map to sites of instability in the Prader- Willi/Angelman syndrome chromosome region (15q11-q13) Hum Mol Genet. 1999;8:1025–1037. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous