

COOH-terminal truncations promote proteasome-dependent degradation of mature cystic fibrosis transmembrane conductance regulator from post-Golgi compartments
- PMID: 11381082
- PMCID: PMC2174331
- DOI: 10.1083/jcb.153.5.957
COOH-terminal truncations promote proteasome-dependent degradation of mature cystic fibrosis transmembrane conductance regulator from post-Golgi compartments
Abstract
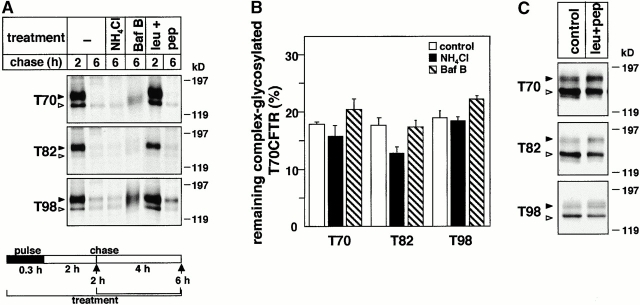
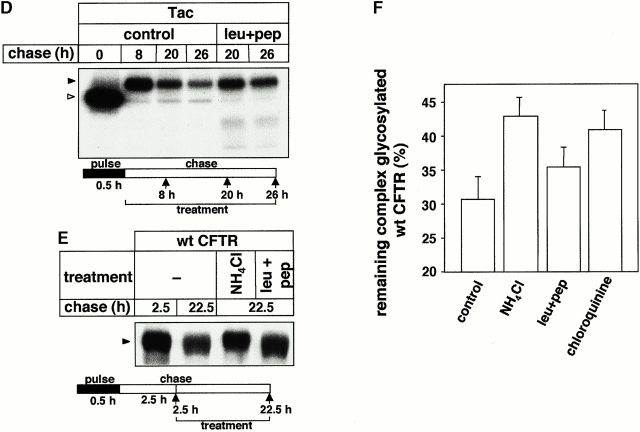
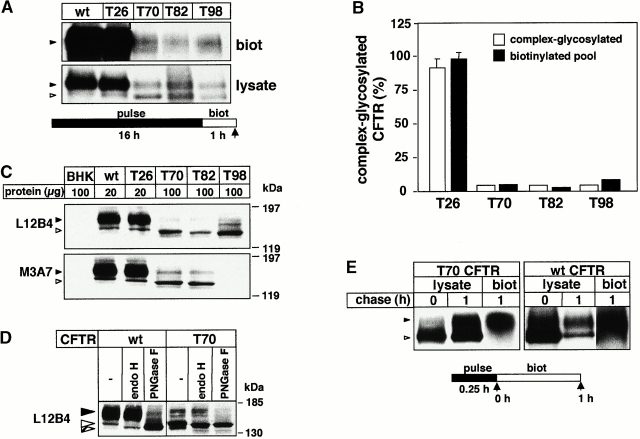
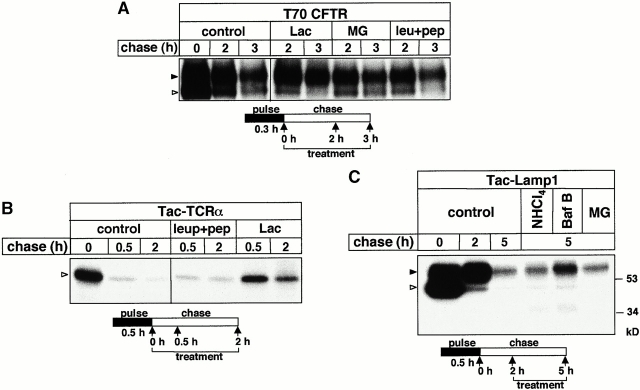
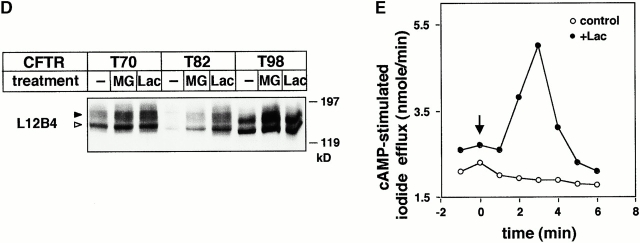
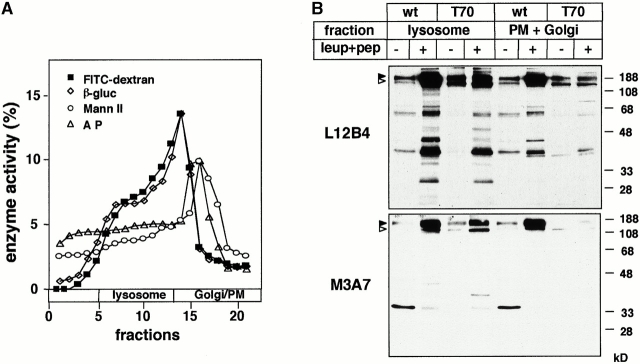
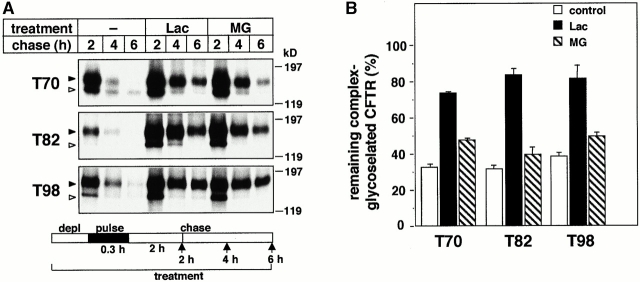
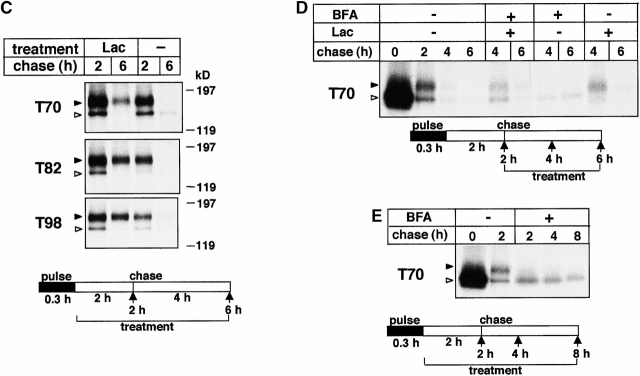
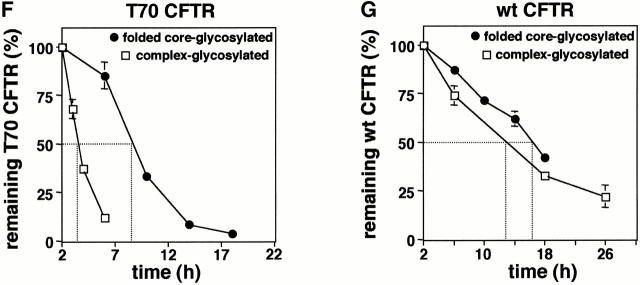
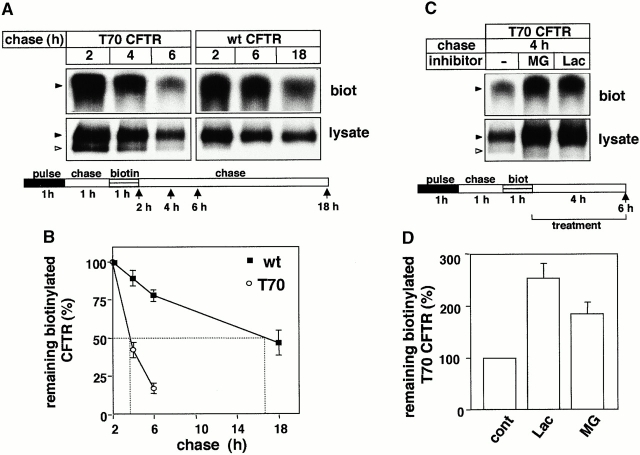
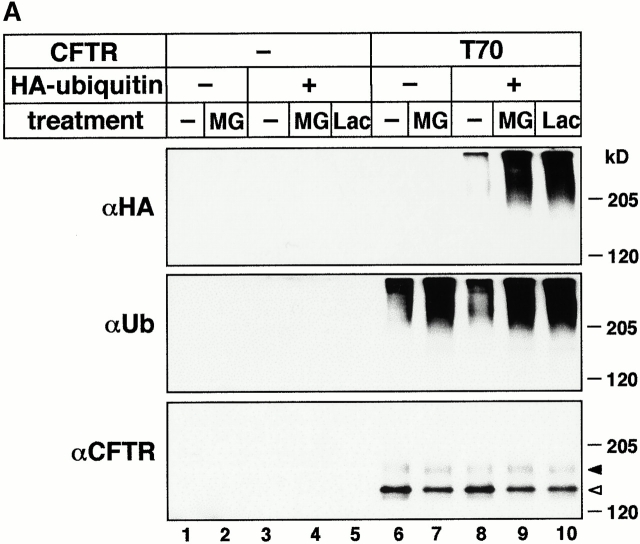
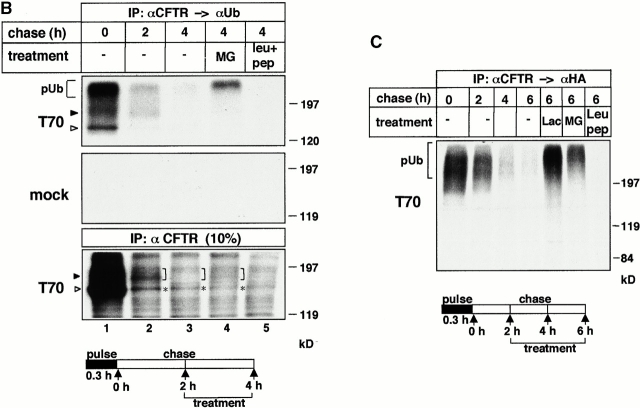
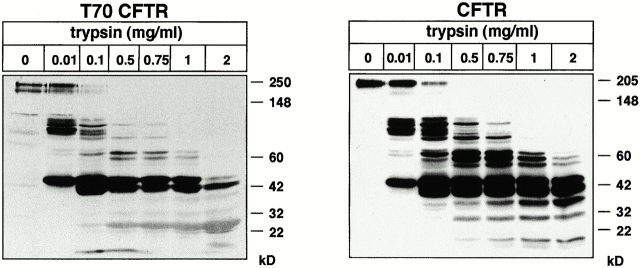
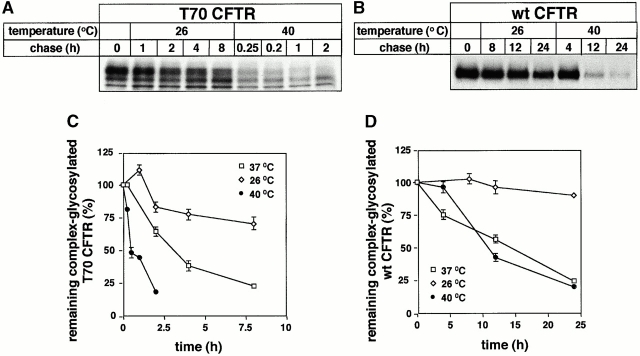
Impaired biosynthetic processing of the cystic fibrosis (CF) transmembrane conductance regulator (CFTR), a cAMP-regulated chloride channel, constitutes the most common cause of CF. Recently, we have identified a distinct category of mutation, caused by premature stop codons and frameshift mutations, which manifests in diminished expression of COOH-terminally truncated CFTR at the cell surface. Although the biosynthetic processing and plasma membrane targeting of truncated CFTRs are preserved, the turnover of the complex-glycosylated mutant is sixfold faster than its wild-type (wt) counterpart. Destabilization of the truncated CFTR coincides with its enhanced susceptibility to proteasome-dependent degradation from post-Golgi compartments globally, and the plasma membrane specifically, determined by pulse-chase analysis in conjunction with cell surface biotinylation. Proteolytic cleavage of the full-length complex-glycosylated wt and degradation intermediates derived from both T70 and wt CFTR requires endolysosomal proteases. The enhanced protease sensitivity in vitro and the decreased thermostability of the complex-glycosylated T70 CFTR in vivo suggest that structural destabilization may account for the increased proteasome susceptibility and the short residence time at the cell surface. These in turn are responsible, at least in part, for the phenotypic manifestation of CF. We propose that the proteasome-ubiquitin pathway may be involved in the peripheral quality control of other, partially unfolded membrane proteins as well.
Figures
References
-
- Aridor M., Balch W.E. Integration of endoplasmic reticulum signaling in health and disease. Nat. Med. 1999;5:745–751. - PubMed
-
- Armstrong J., Patel S., Riddle P. Lysosomal sorting mutants of coronavirus E1 protein, a Golgi membane protein. J. Cell Sci. 1990;95:191–197. - PubMed
-
- Baumeister W., Walz J., Zuhi F., Seemuller E. The proteasomeparadigm of a self-compartmentalizing protease. Cell. 1998;92:367–380. - PubMed
