

Ezrin is a downstream effector of trafficking PKC-integrin complexes involved in the control of cell motility
- PMID: 11387207
- PMCID: PMC125254
- DOI: 10.1093/emboj/20.11.2723
Ezrin is a downstream effector of trafficking PKC-integrin complexes involved in the control of cell motility
Abstract
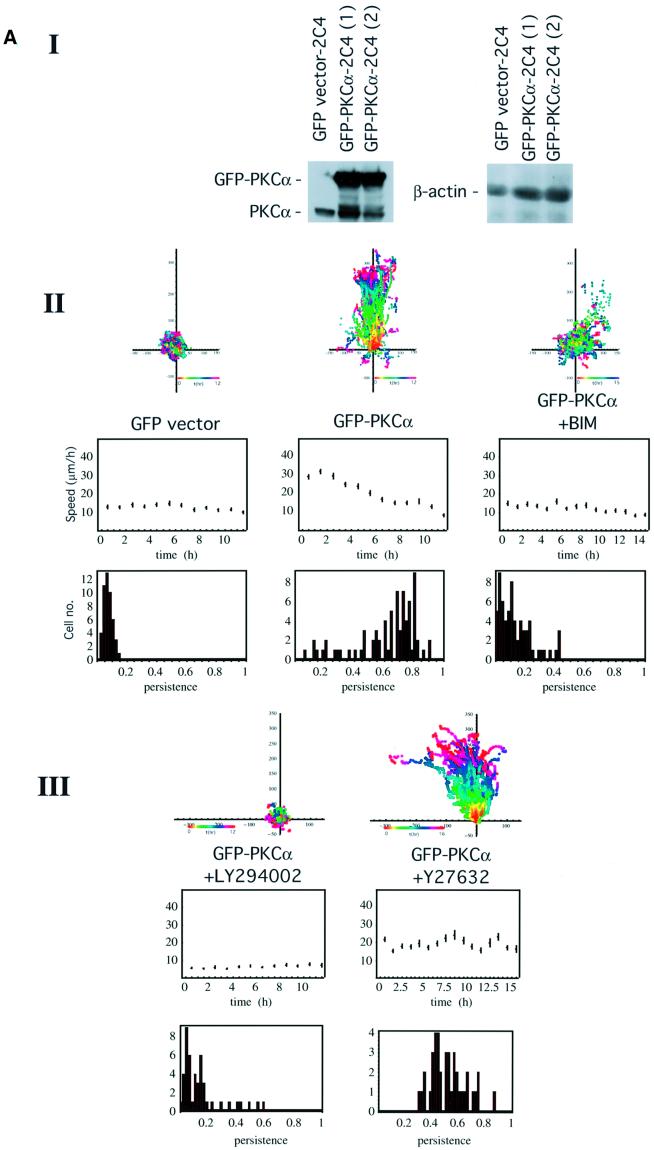
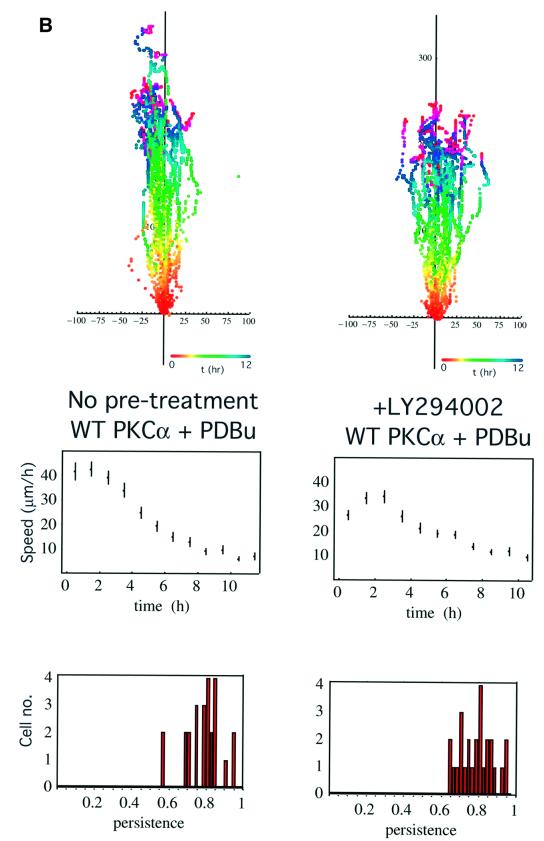
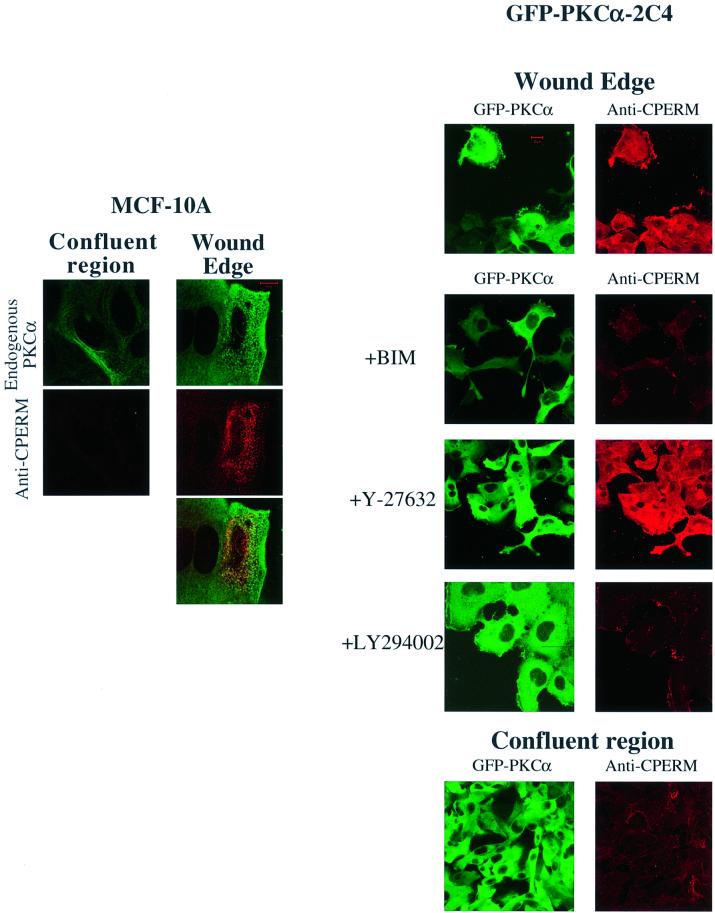
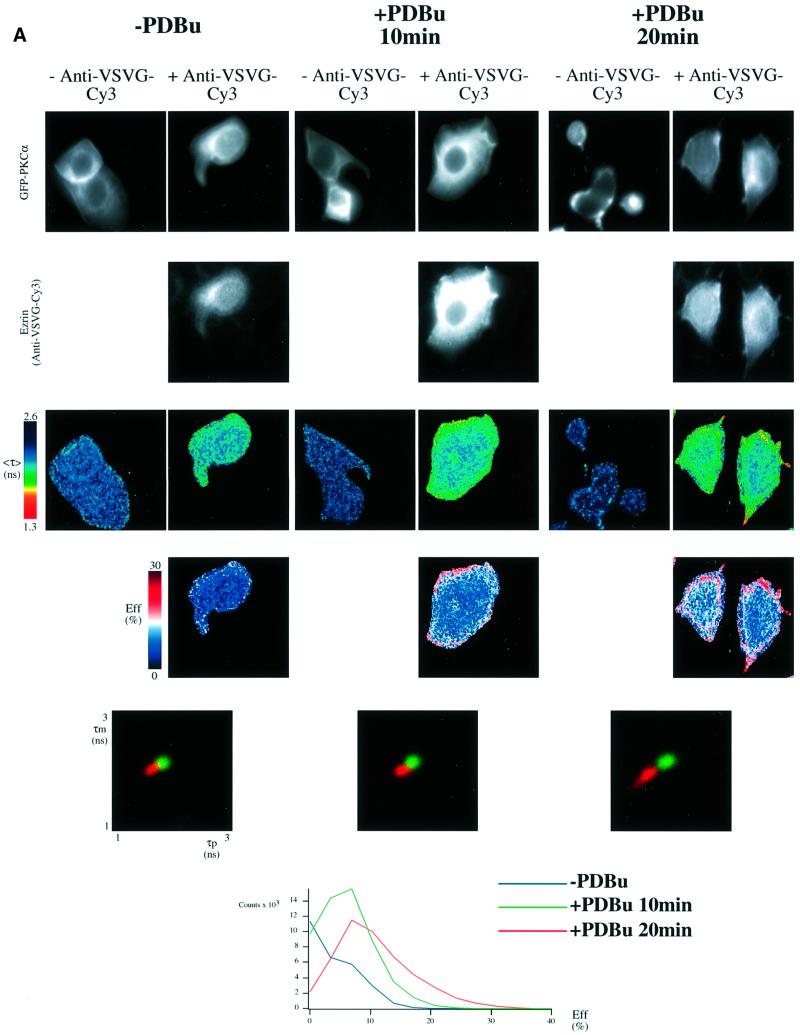
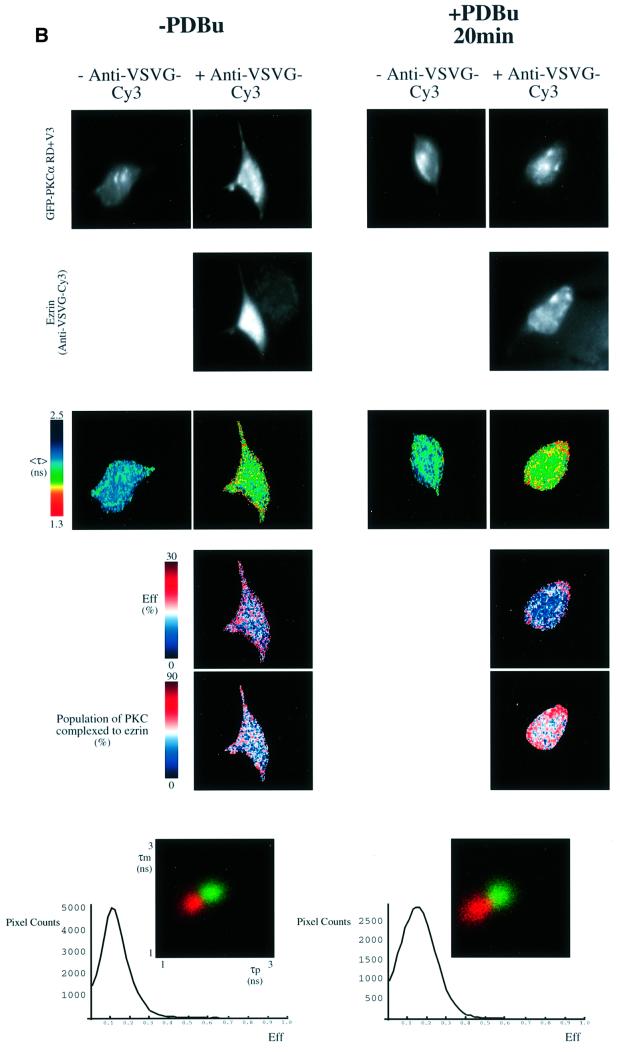
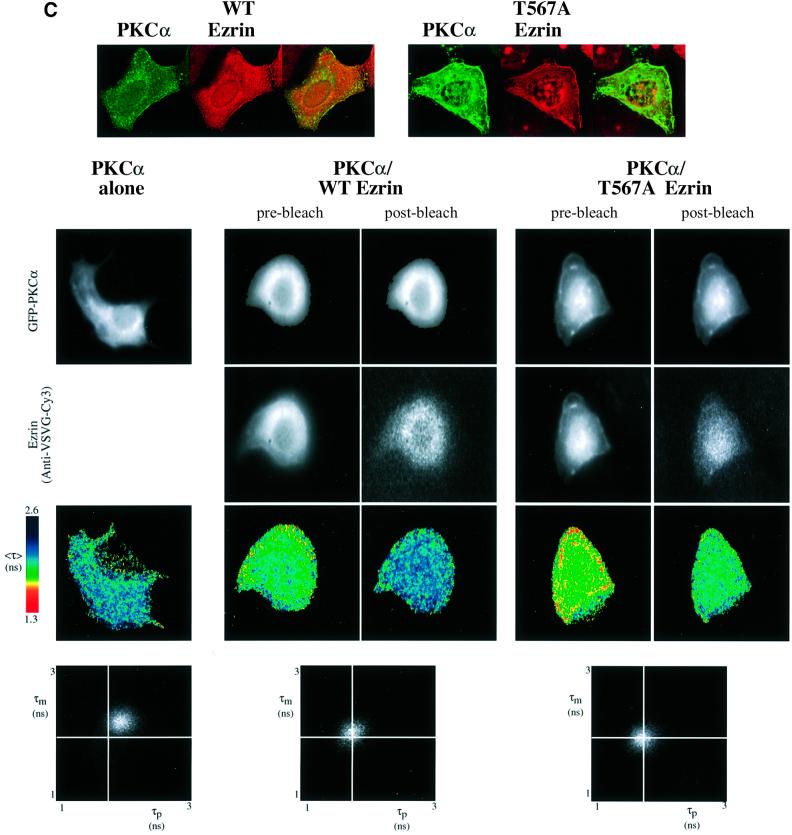
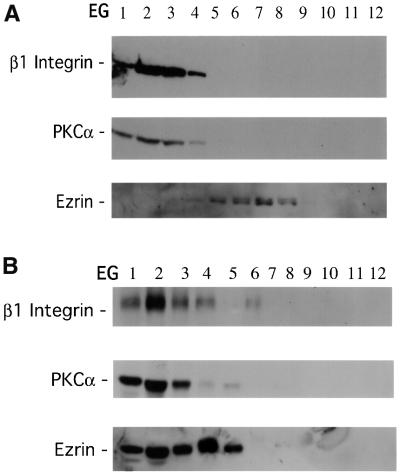
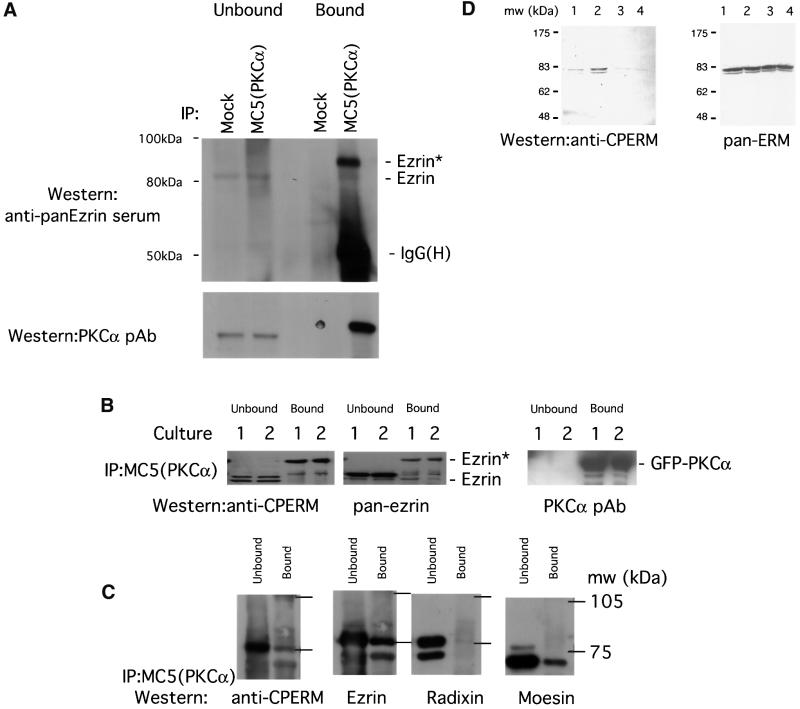
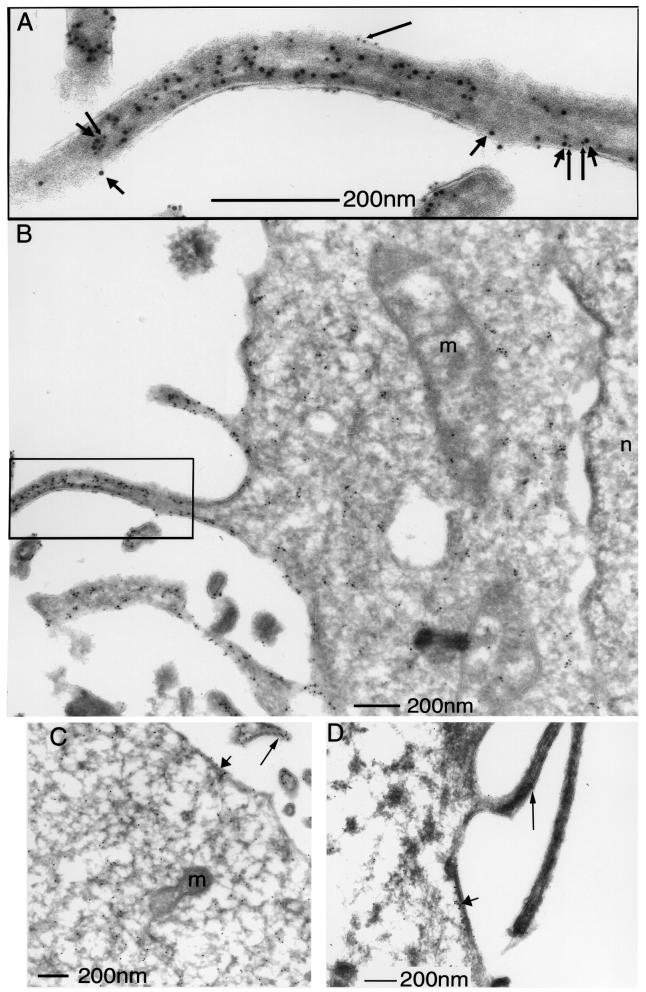
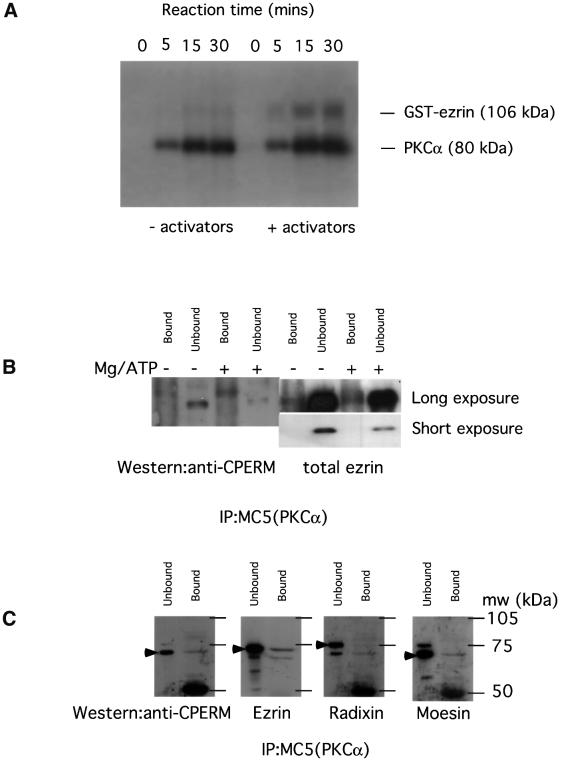
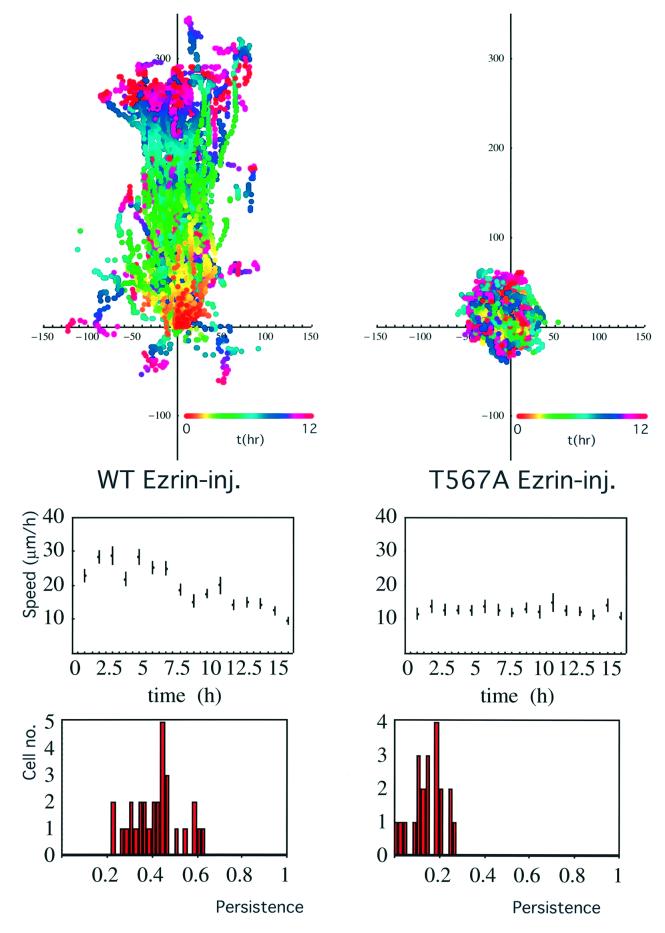
Protein kinase C (PKC) alpha has been implicated in beta1 integrin-mediated cell migration. Stable expression of PKCalpha is shown here to enhance wound closure. This PKC-driven migratory response directly correlates with increased C-terminal threonine phosphorylation of ezrin/moesin/radixin (ERM) at the wound edge. Both the wound migratory response and ERM phosphorylation are dependent upon the catalytic function of PKC and are susceptible to inhibition by phosphatidylinositol 3-kinase blockade. Upon phorbol 12,13-dibutyrate stimulation, green fluorescent protein-PKCalpha and beta1 integrins co-sediment with ERM proteins in low-density sucrose gradient fractions that are enriched in transferrin receptors. Using fluorescence lifetime imaging microscopy, PKCalpha is shown to form a molecular complex with ezrin, and the PKC-co-precipitated endogenous ERM is hyperphosphorylated at the C-terminal threonine residue, i.e. activated. Electron microscopy showed an enrichment of both proteins in plasma membrane protrusions. Finally, overexpression of the C-terminal threonine phosphorylation site mutant of ezrin has a dominant inhibitory effect on PKCalpha-induced cell migration. We provide the first evidence that PKCalpha or a PKCalpha-associated serine/threonine kinase can phosphorylate the ERM C-terminal threonine residue within a kinase-ezrin molecular complex in vivo.
Figures
References
-
- Arber S., Barbayannis,F.A., Hanser,H., Schneider,C., Stanyon,C.A., Bernard,O. and Caroni,P. (1998) Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature, 393, 805–809. - PubMed
-
- Bae Y.S., Cantley,L.G., Chen,C.S., Kim,S.R., Kwon,K.S. and Rhee,S.G. (1998) Activation of phospholipase C-γ by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem., 273, 4465–4469. - PubMed
-
- Bastiaens P.I.H. and Jovin,T.M. (1998) Fluorescence resonance energy transfer microscopy. In Celis,J. (ed.), Cell Biology: A Laboratory Handbook. Academic Press, New York, NY, pp. 136–146.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
