

Ampa/kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter
- PMID: 11404409
- PMCID: PMC6762765
- DOI: 10.1523/JNEUROSCI.21-12-04237.2001
Ampa/kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter
Abstract
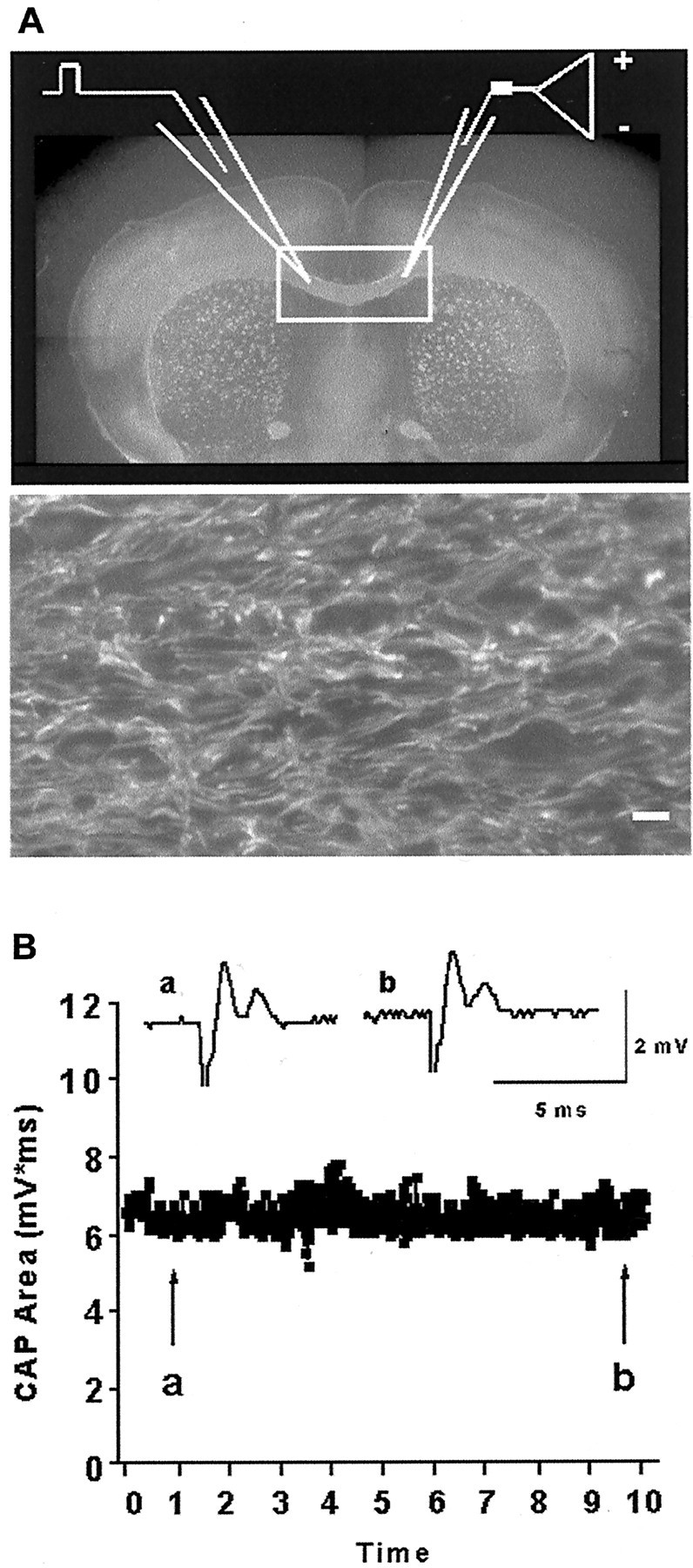
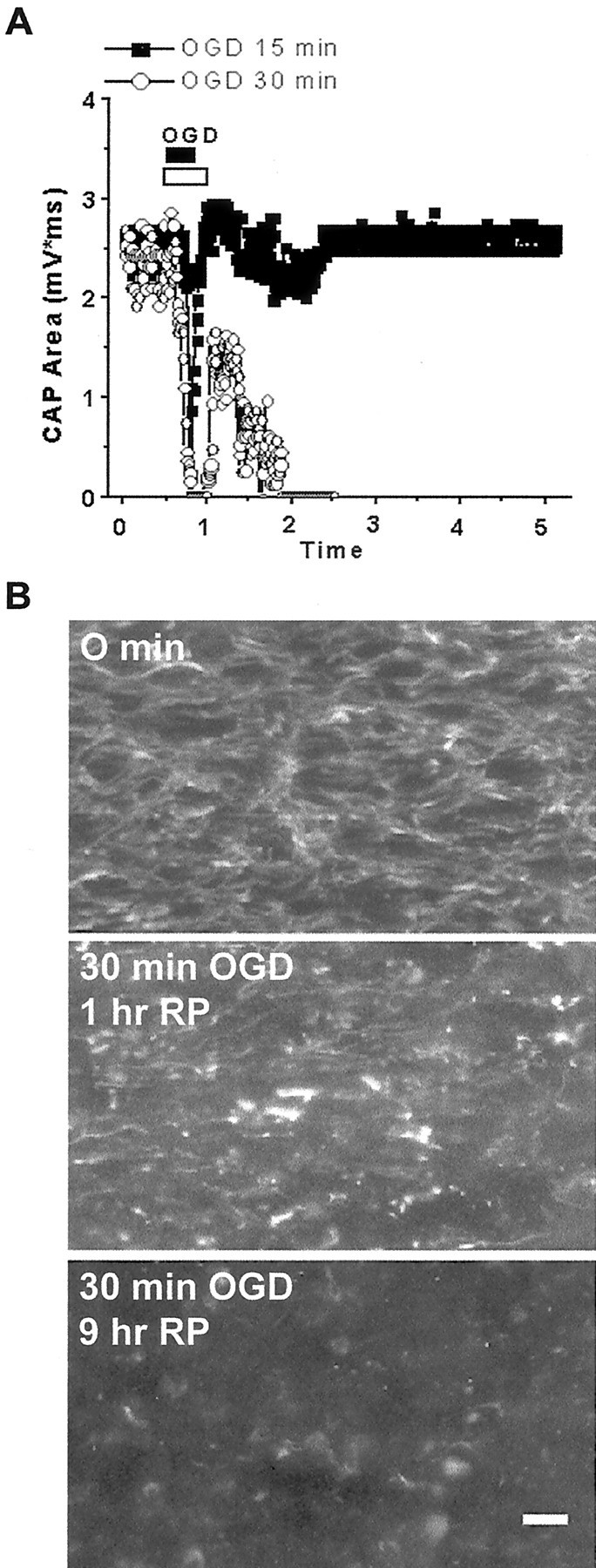
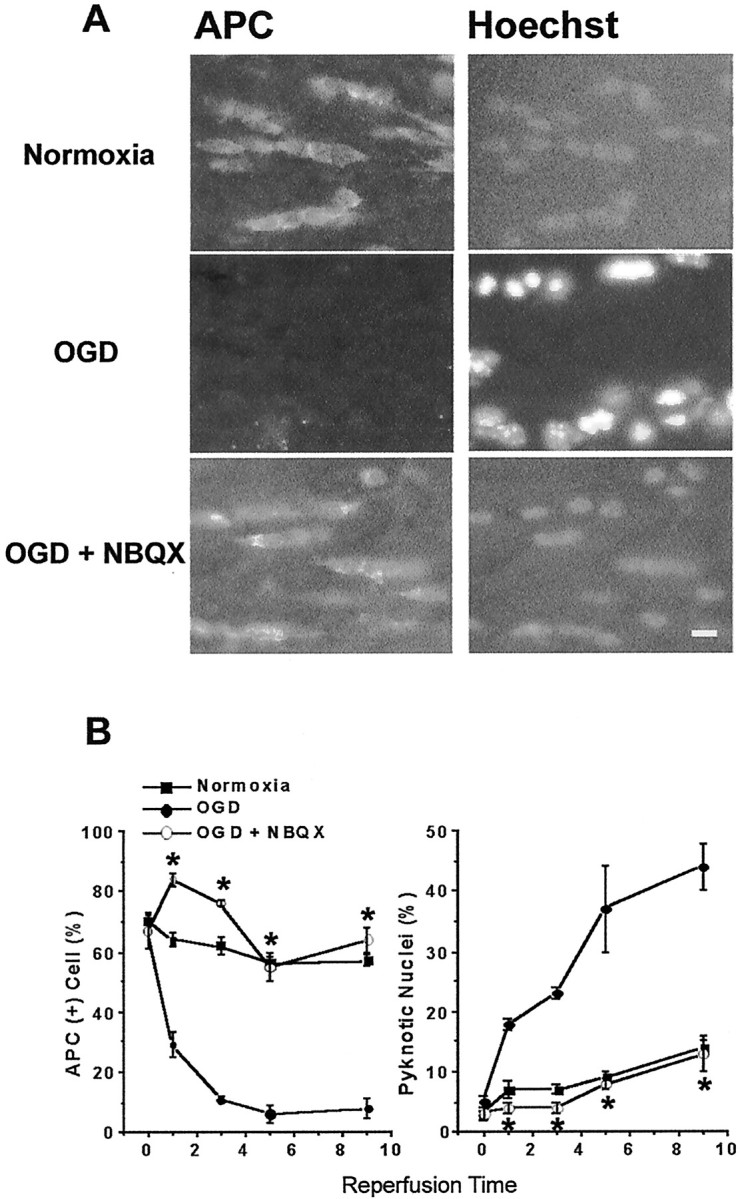
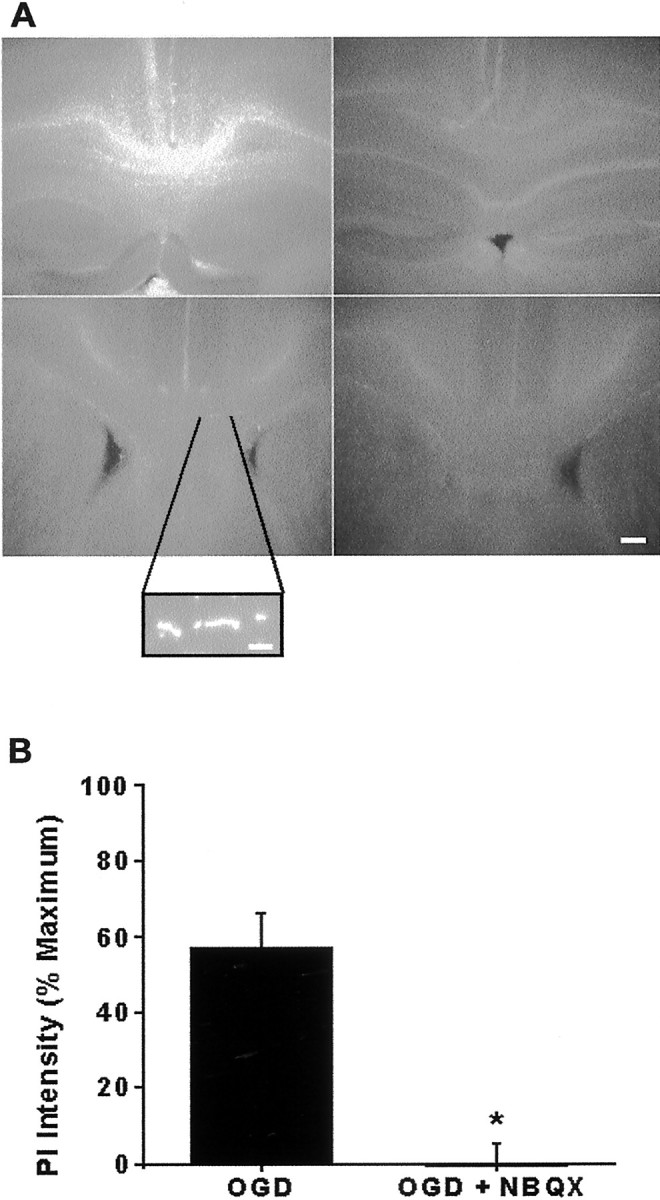
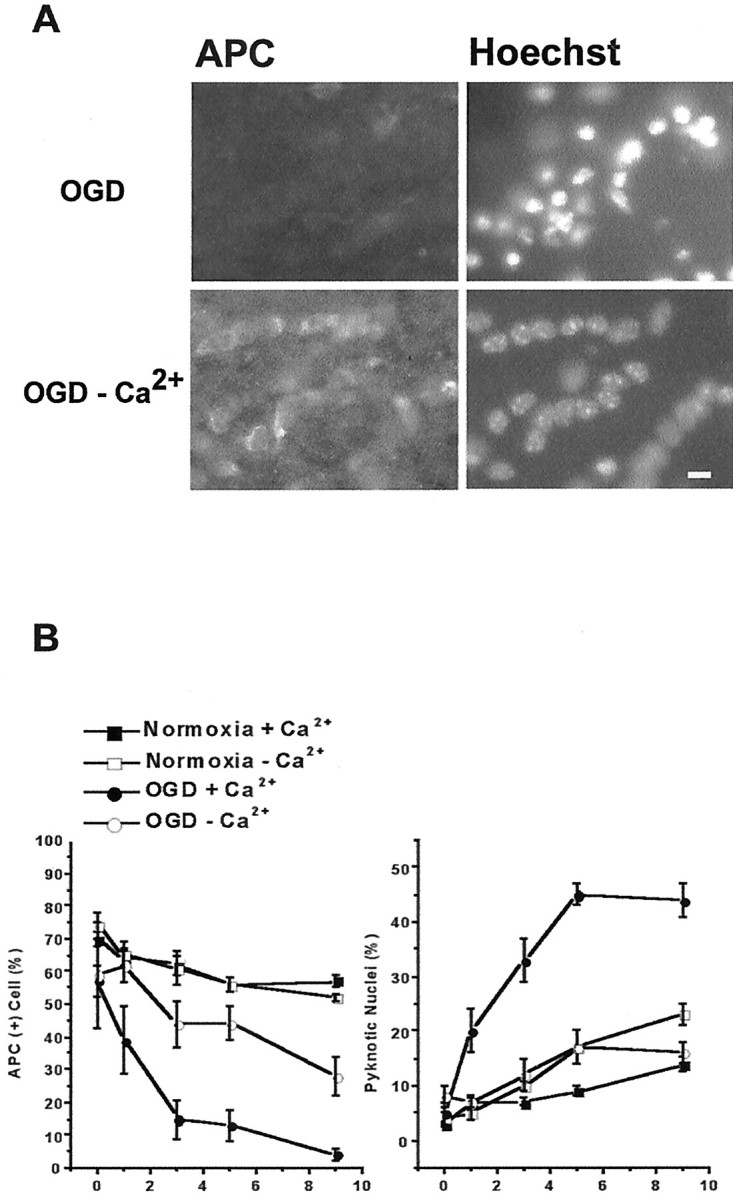
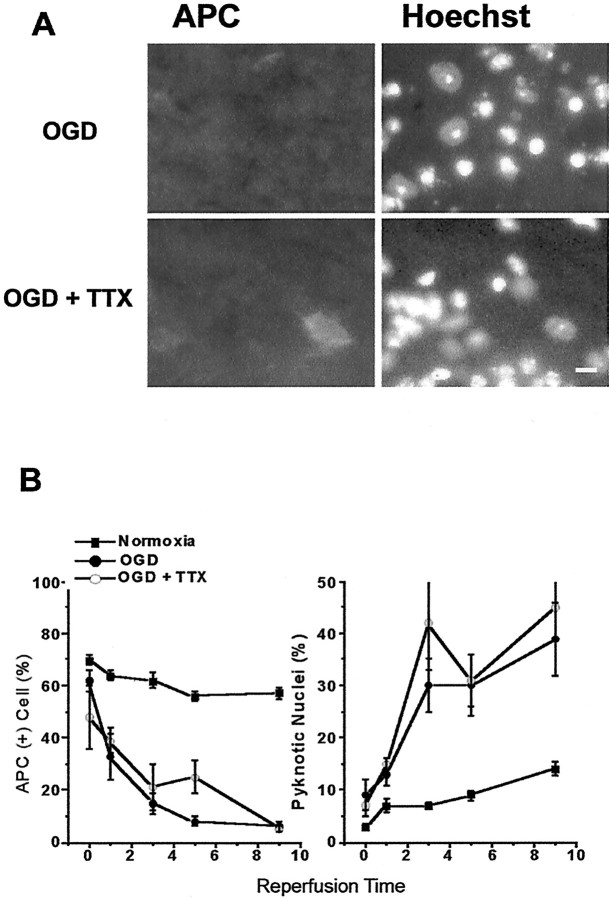
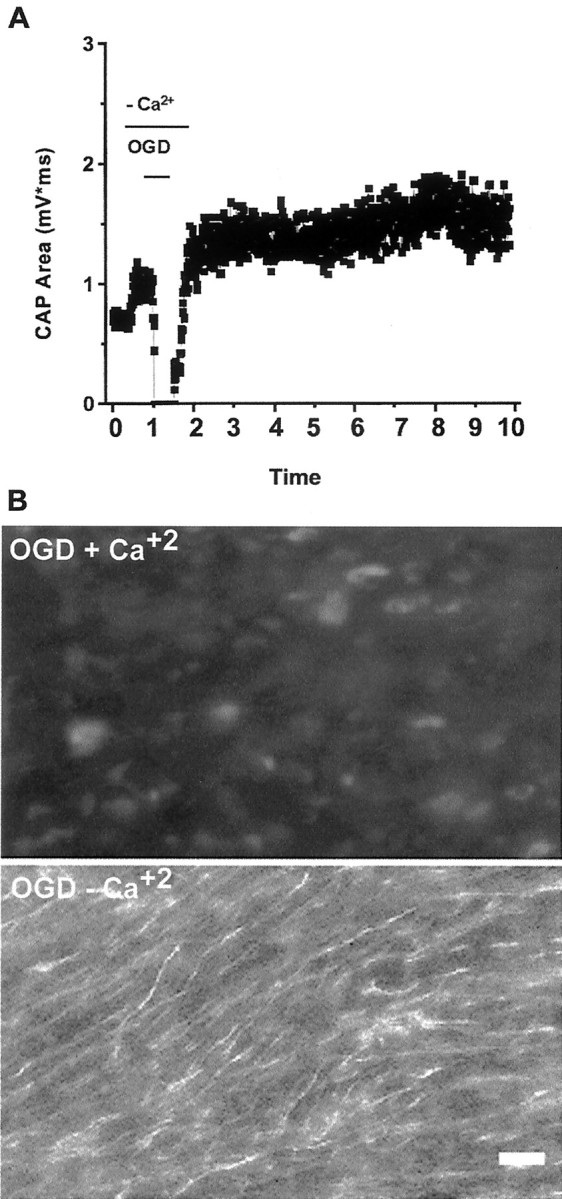
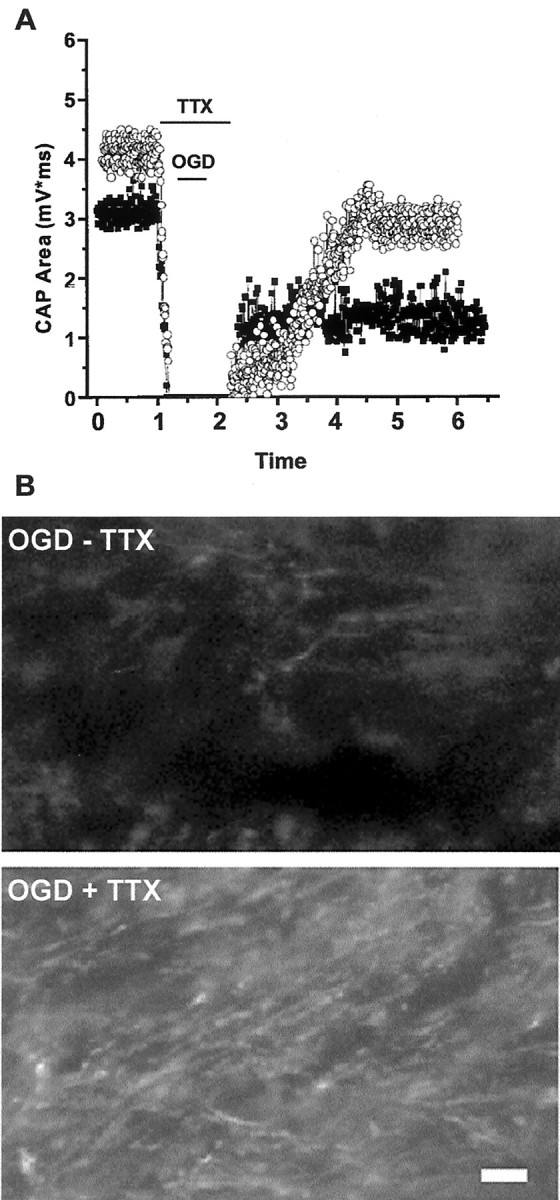
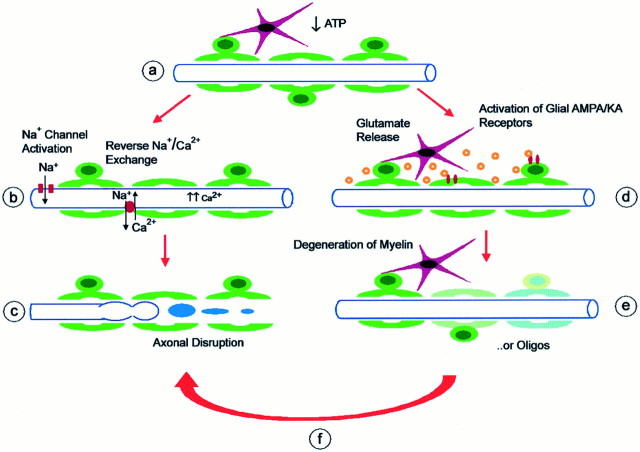
We developed an in situ model to investigate the hypothesis that AMPA/kainate (AMPA/KA) receptor activation contributes to hypoxic-ischemic white matter injury in the adult brain. Acute coronal brain slices, including corpus callosum, were prepared from adult mice. After exposure to transient oxygen and glucose deprivation (OGD), white matter injury was assessed by electrophysiology and immunofluorescence for oligodendrocytes and axonal neurofilaments. White matter cellular components and the stimulus-evoked compound action potential (CAP) remained stable for 12 hr after preparation. OGD for 30 min resulted in an irreversible loss of the CAP as well as structural disruption of axons and subsequent loss of neurofilament immunofluorescence. OGD also caused widespread oligodendrocyte death, demonstrated by the loss of APC labeling and the gain of pyknotic nuclear morphology and propidium iodide labeling. Blockade of AMPA/KA receptors with 30 microm NBQX or the AMPA-selective antagonist 30 microm GYKI 52466 prevented OGD-induced oligodendrocyte death. Oligodendrocytes also were preserved by the removal of Ca(2+), but not by a blockade of voltage-gated Na(+) channels. The protective action of NBQX was still present in isolated corpus callosum slices. CAP areas and axonal structure were preserved by Ca(2+) removal and partially protected by a blockade of voltage-gated Na(+) channels. NBQX prevented OGD-induced CAP loss and preserved axonal structure. These observations highlight convergent pathways leading to hypoxic-ischemic damage of cerebral white matter. In accordance with previous suggestions, the activation of voltage-gated Na(+) channels contributes to axonal damage. Overactivation of glial AMPA/KA receptors leads to oligodendrocyte death and also plays an important role in structural and functional disruption of axons.
Figures


References
-
- Adams JH, Graham DI, Gennarelli TA. Head injury in man and experimental animals: neuropathology. Acta Neurochir Suppl (Wien) 1983;32:15–30. - PubMed
-
- Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. - PubMed
-
- Baltan Tekkök S, Hyrc KL, Underhill SM, Goldberg MP. Na+/Ca2+ exchange blocker KB-R7943 protects axons and oligodendrocytes during oxygen glucose deprivation. Soc Neurosci Abstr. 2000;26:2065.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous